
Introduction
MetamORF aims to provide a repository of unique short Open Reading Frames (sORFs) identified in the human and mouse genomes with both experimental and computational approaches.
By gathering publicly available sORF data, normalizing it and summarizing redundant information, 2,594,154 unique sORFs have been registered in the database. The data has been re-processed in order to propose a normalized nomenclature, in particular of ORF categories.
| Feature | Homo sapiens | Mus musculus | Total | |
|---|---|---|---|---|
| Data sources | ORFs | 1,344,978 | 1,249,176 | 2,594,154 |
| Transcripts | 101,597 | 85,653 | 187,250 | |
| ORF to transcript associations | 3,379,219 | 2,066,627 | 5,445,846 | |
| MetamORF | Unique ORFs | 664,771 | 497,904 | 1,162,675 |
| Unique transcripts | 90,406 | 63,147 | 153,553 | |
| Unique ORF to transcript associations | 729,793 | 696,785 | 1,426,578 | |
MetamORF web interface allows to easily navigate through the database, to find sORFs located on particular genes, transcripts or locus of interest. It helps you browsing the data, visualizing and filtering it using multiple criteria (such as ORF lengths, annotations, cell types...) and exporting the results at several convenient formats. In addition to the current web interface, the database content is also available through track hubs at UCSC Genome Browser. Links to these genome tracks and from UCSC genome tracks to MetamORF website have been implemented to help you navigate from one to another.
For extensive information regarding the criteria of data sources inclusion we use and the processing of data, please see the publications related to the MetamORF database as well as the dedicated section of the current documentation.
Tutorial
Browsing the database and searching for ORFs
The MetamORF web interface allows you either to browse the full database for one species, browse all the data related to a particular cell type or to start your search from:
- A gene symbol, ID, name or alias (NBCI and HGNC gene IDs respectively needs to be preceed by the NCBI: and HGNC: prefixes). Examples: ATF4, HGNC:19687, ENSG00000172071, NCBI:23645...
- A transcript ID or name. Examples: ENST00000003912, CD9-201, CYP4F2-201...
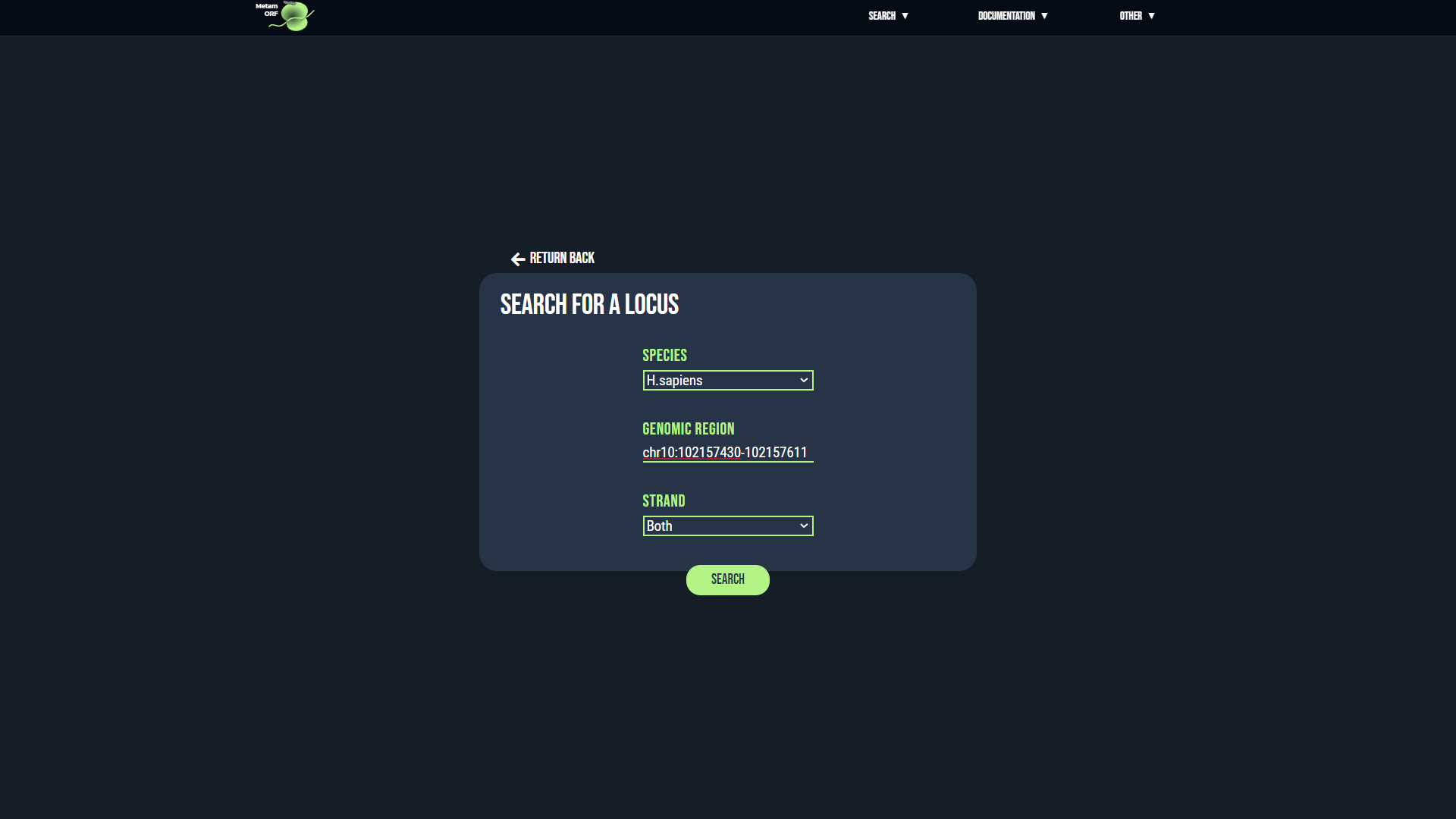
- A locus, provided at the format chrName:start-end, where chrName is the chromosome name (or scaffold), preceed by the 'chr' prefix, start is the lower value of the genomic region you are looking for and end value is the upper value of the genomic region (in a 1-based coordinates system). Example: chr18:165,583-265,582...
- A MetamORF ORF ID.
Searching for ORFs
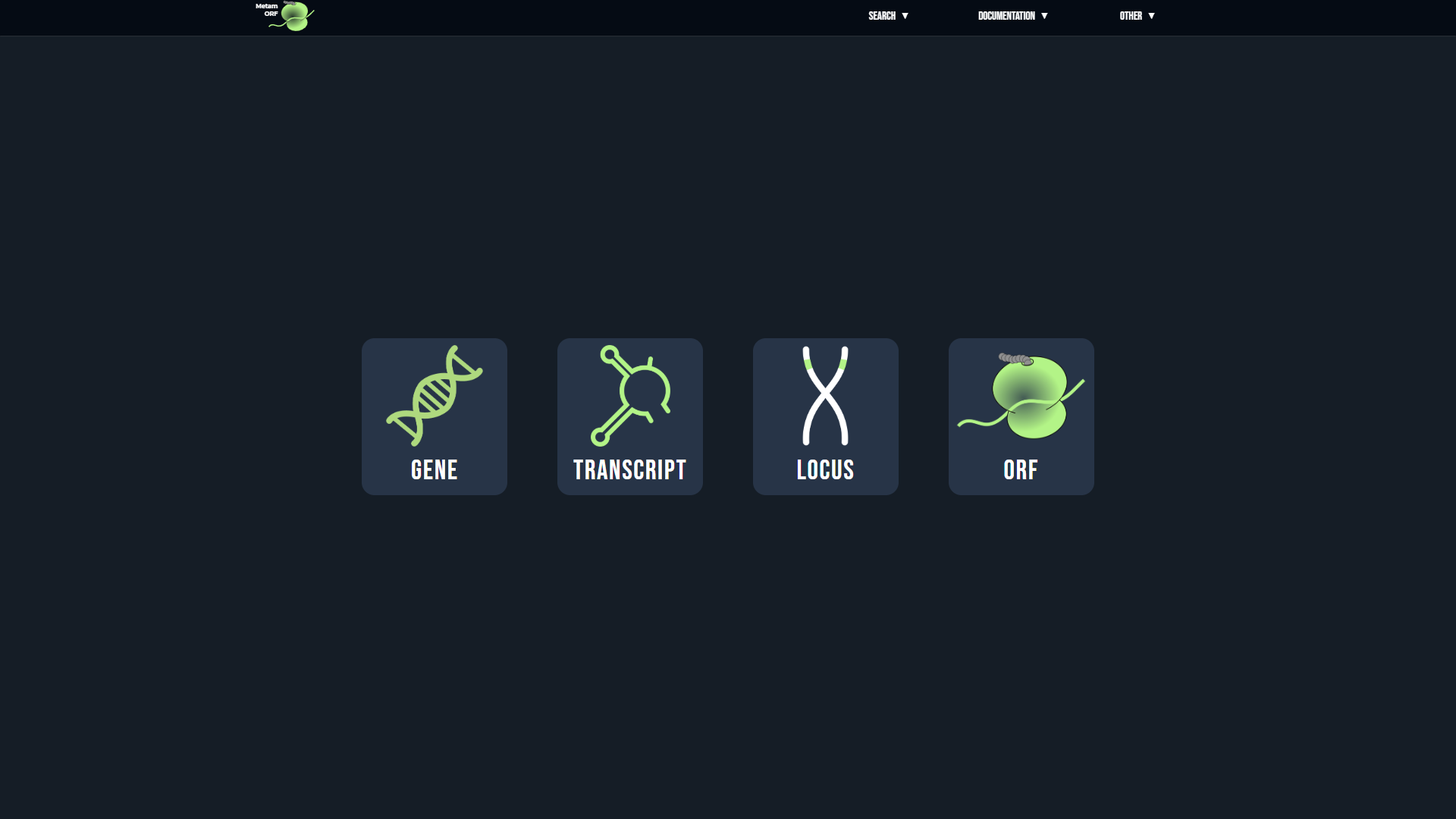
Once you have selected your search mode, you will be asked to select the species you are interested in (the database includes information for H. sapiens and M. musculus).
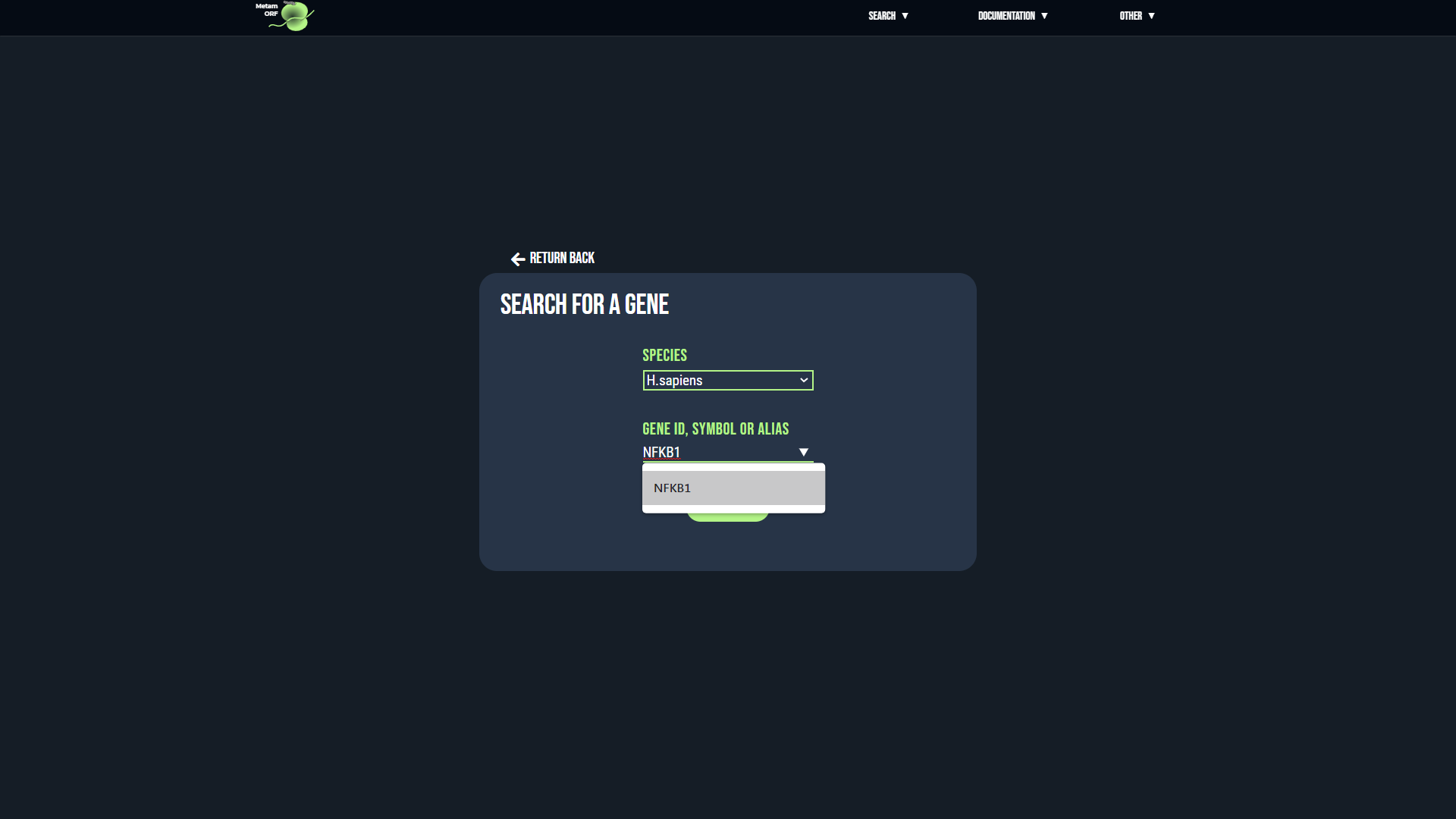
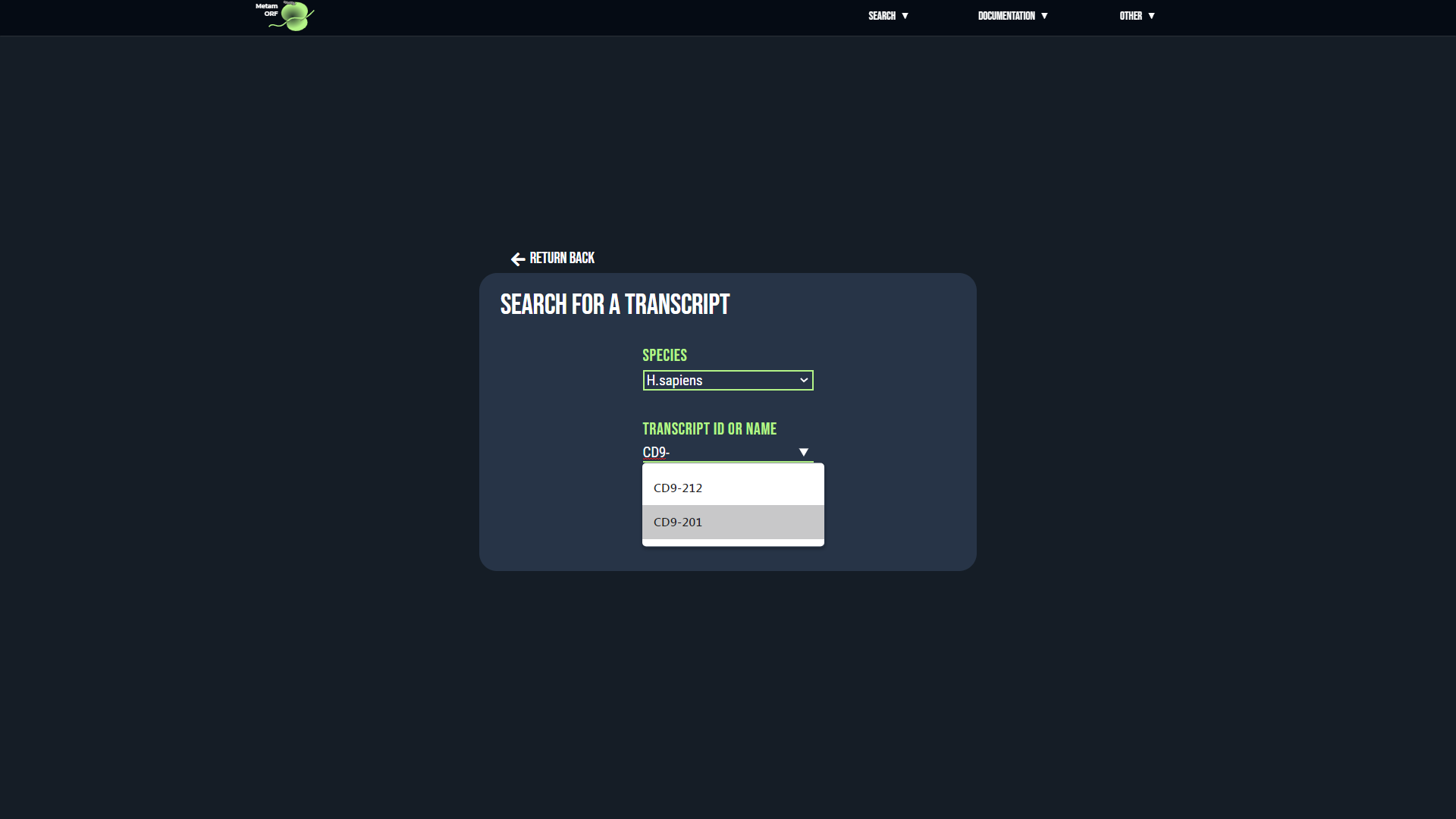
For gene and transcript searchs, if there is an ambiguity regarding the reference provided, you will be directed to an intermediate page asking you to precise your choice. This may happen for instance when the same alias is used by several genes.
- In case of ambiguity on the gene searched, for each gene that matches your request, its ID as well as the list of its known aliases and the name of its chromosome (or scaffold) will be displayed to help you make your selection. Once you have identified the gene you are looking for, click on its ID to be redirected to the page of the gene.
- In case of ambiguity on the transcript searched, for each transcript that matches your request, its MetamORF ID, external ID, name, strand, biotype, related gene ID and aliases, start and end positions as well as CDS start and stop positions (for coding transcripts) will be displayed to help you make your selection. Once you have identified the transcript you are looking for, click on its MetamORF ID to be redirected to the page of the transcript.
Examples of pages displayed in case of ambiguity
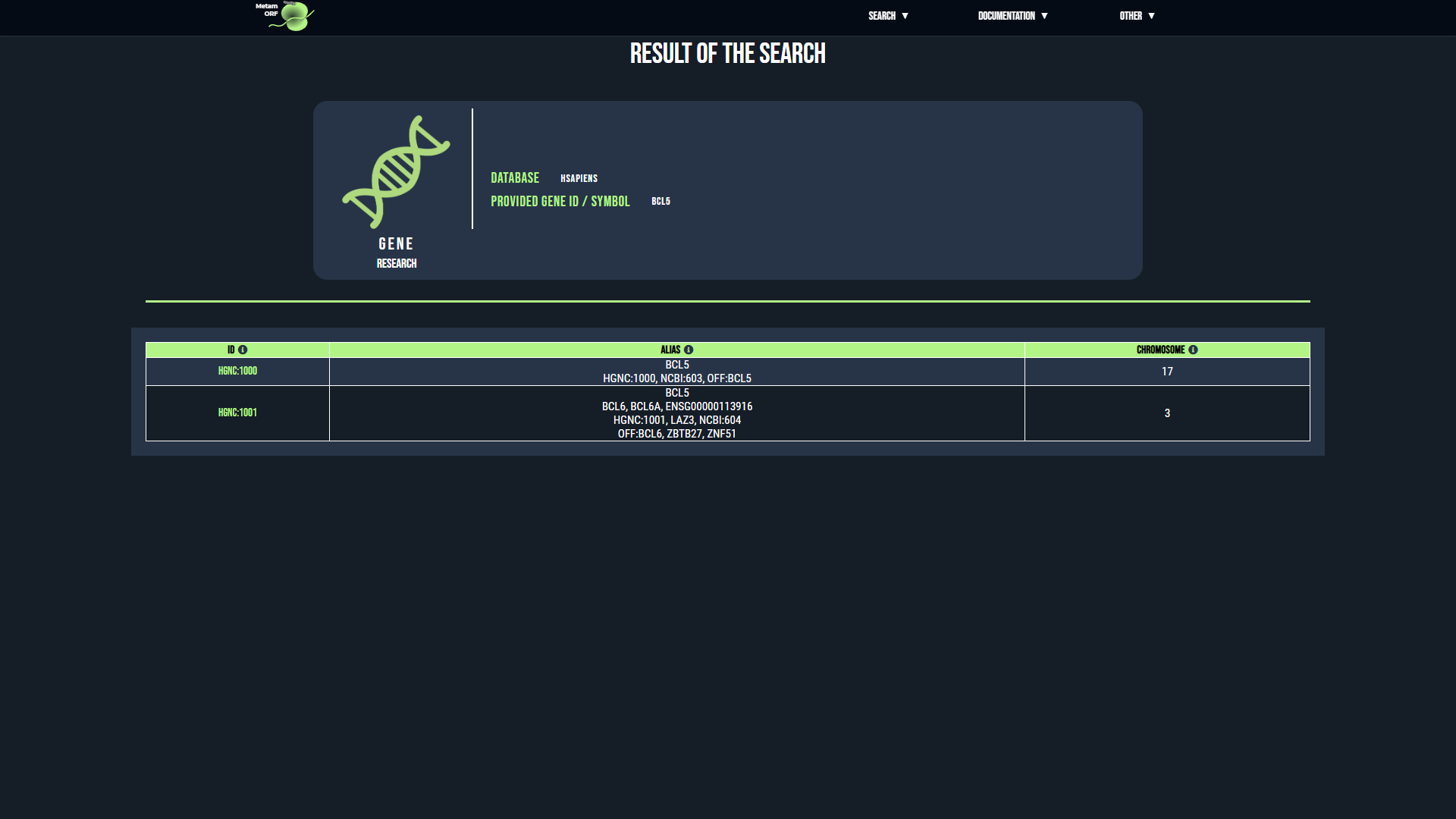
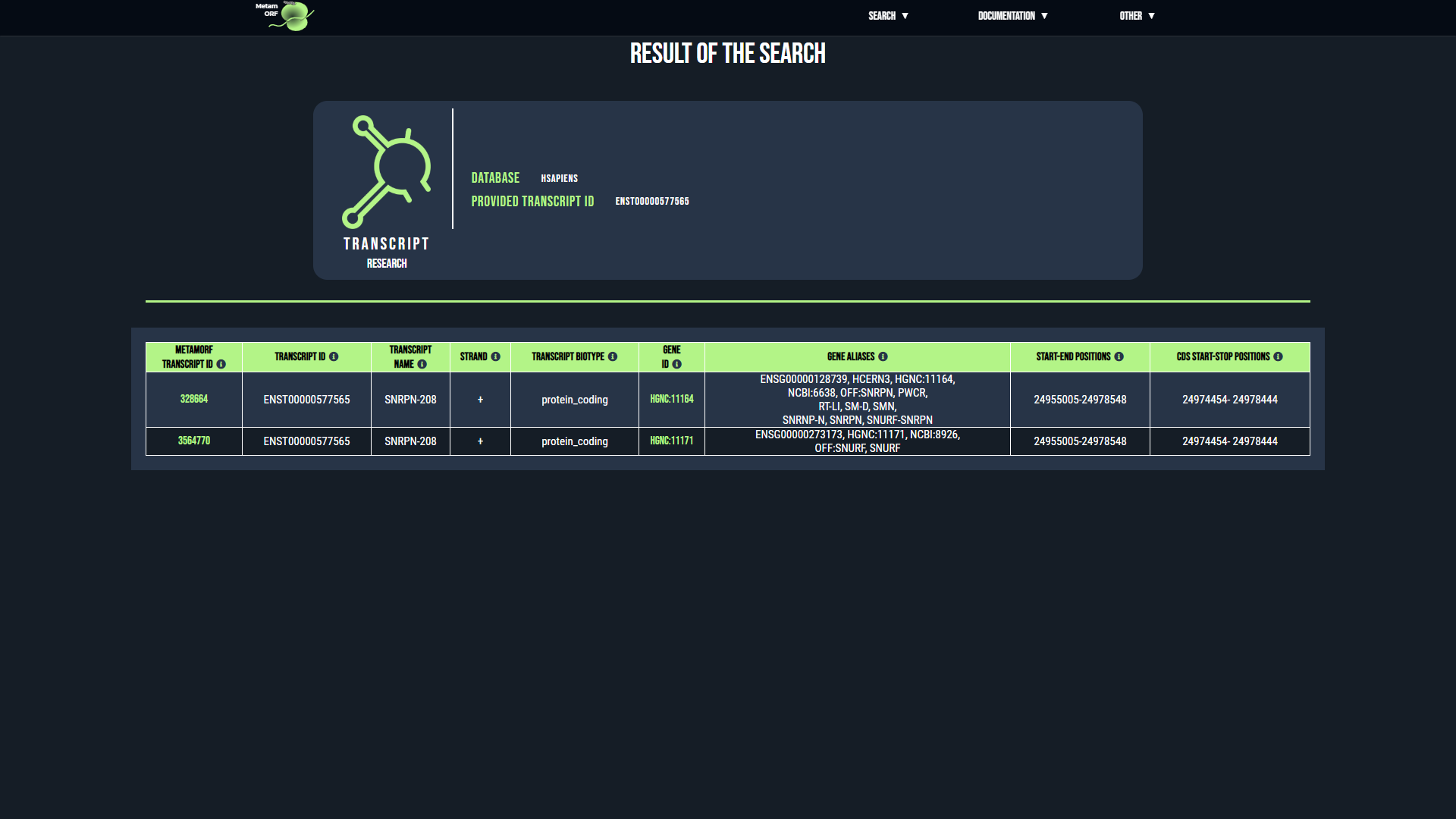
Additional information:
- Your research may not succeed if the reference you provided (i.e. the gene or transcript ID / symbol / name / alias) is missing from our database. In such case, you research will failed and will be redirected to the search page.
- If you are directed to a page that does not contains any ORF, this means that the gene, transcript or locus you are looking for is well registered in our database but there is not any ORF related to the gene or transcript or located in the locus.
- We suggest to consult Ensembl, NCBI, HUGO and MGI resources to help you find other references (ID, symbol, name, alias) of the gene or transcript you are looking for and to make again your request with one of its alternative reference.
- Please note that some transcript cross-references are missing from the MetamORF database. In particular transcripts that do not harbor any ORF are not registered. Hence, if your search do not return any result for a particular transcript, we strongly advice to perform a new search using the gene related to your transcript of interest.
- Please note also that genomic region larger than 5,000,000 nucleotides are not allowed as they are susceptible to return too many results. If you need to perform queries on larger region, please contact us.
Interpreting the results
Once the search performed you will be able to navigate the database through five different types of pages:
- The browser
- A gene page (or "gene-centric view", default result page from gene search)
- A transcript page (or "transcript-centric view", default result page from transcript search)
- An ORF page (or "ORF-centric view")
- A locus page (default result page from locus search)
Each of these page is decribed below.
Browser
The browser allows to get information about all the ORFs registered in MetamORF and matching the filters you selected (no filters applied by default). The first section of the page remind the filters selected whilst the second describes all the ORFs matching your request.
Example of result in the browser
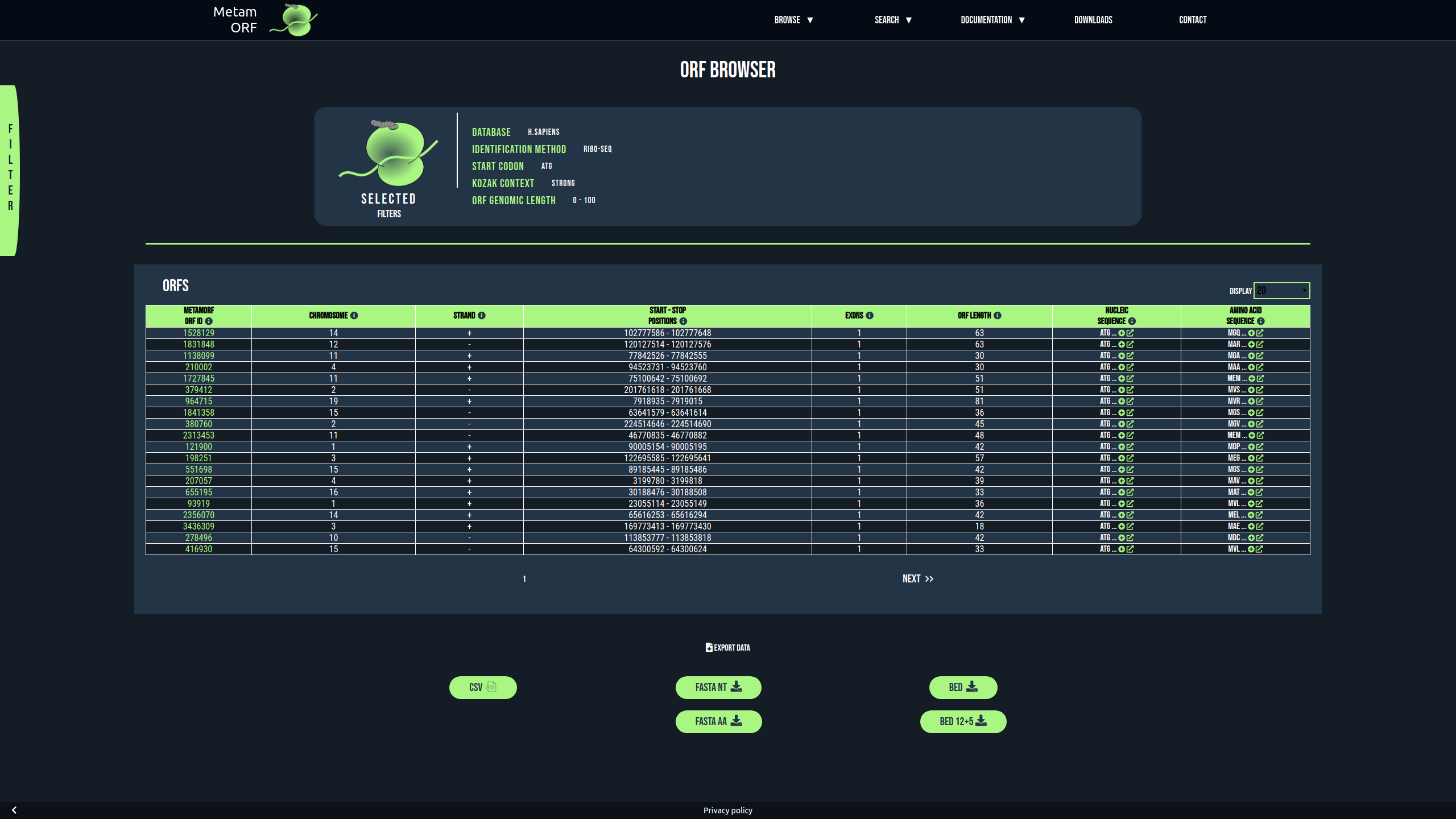
The rows registering ORFs contain the following columns:
- MetamORF ORF ID: The MetamORF ID of the ORF. This is an unique ID referring to the ORF. You may click on it to access to the ORF centric view corresponding to the entry.
- Chromosome: The name of the chromosome (or scaffold) the ORF is located on. You may click on it to access to the ORF centric view corresponding to the entry.
- Strand: The strand of the ORF (+ or -)
- Start - stop positions: The absolute genomic positions (i.e. the positions on the chromosome) of the start and stop codons of the ORF (in a 1-based system).
- Exons: The number of exons constituting the ORF
- ORF length: The genomic length of the ORF (in bp). This length is defined as the sum of the lengths of each exon constituting the ORF (thus excluding its eventual introns) and includes both the start and the stop codons.
- Nucleic sequence: The nucleic sequence of the ORF. Only the three first nucleotids are displayed, click on the button to display the full sequence in a pop-up or on the button to display the full sequence in a fasta-like format in a new window.
- Amino acid sequence: The amino acid sequence of the ORF. Only the three first amino acids are displayed, click on the button to display the full sequence in a pop-up or on the button to display the full sequence in a fasta-like format in a new window.
As this page allows to display a large number of entries (all the ORFs registered in MetamORF for a species at most), the most important information related to the ORF are displayed. Additional details related to these ORFs such as their annotations, the cell contexts in which they have been identified or the name of the original data sources that described them, may be found in the ORF page. In addition, only 50 results are displayed on the page by default. This value can be changed by setting the Display parameter to another value (5,10,20,50,100 or 200). The Next and Previous buttons allow to navigate through the results.
Gene page
From this page, you will be able to get information regarding the ORFs and transcripts which are related to a particular gene. The first section describes the gene itself whilst the second describes its transcripts and its ORFs.
By default, the results are grouped by ORFs, meaning that the table of results contains one unique row for each ORF that is related to the gene. For each of these ORFs you may expand the table to get information regarding the transcripts that harbor the ORF (use the button located in the ORFs column to do so).
You may also group the results by transcript. In such cases, the table of results contains one unique row for each transcript that is related to the gene. For each of these transcripts you may expand the table to get information regarding the ORFs it harbors (use the button located in the Transcripts column to do so).
The attributes registered in the tables are the same, whatever the group method chosen is.
Group the results by ORFs or by transcripts
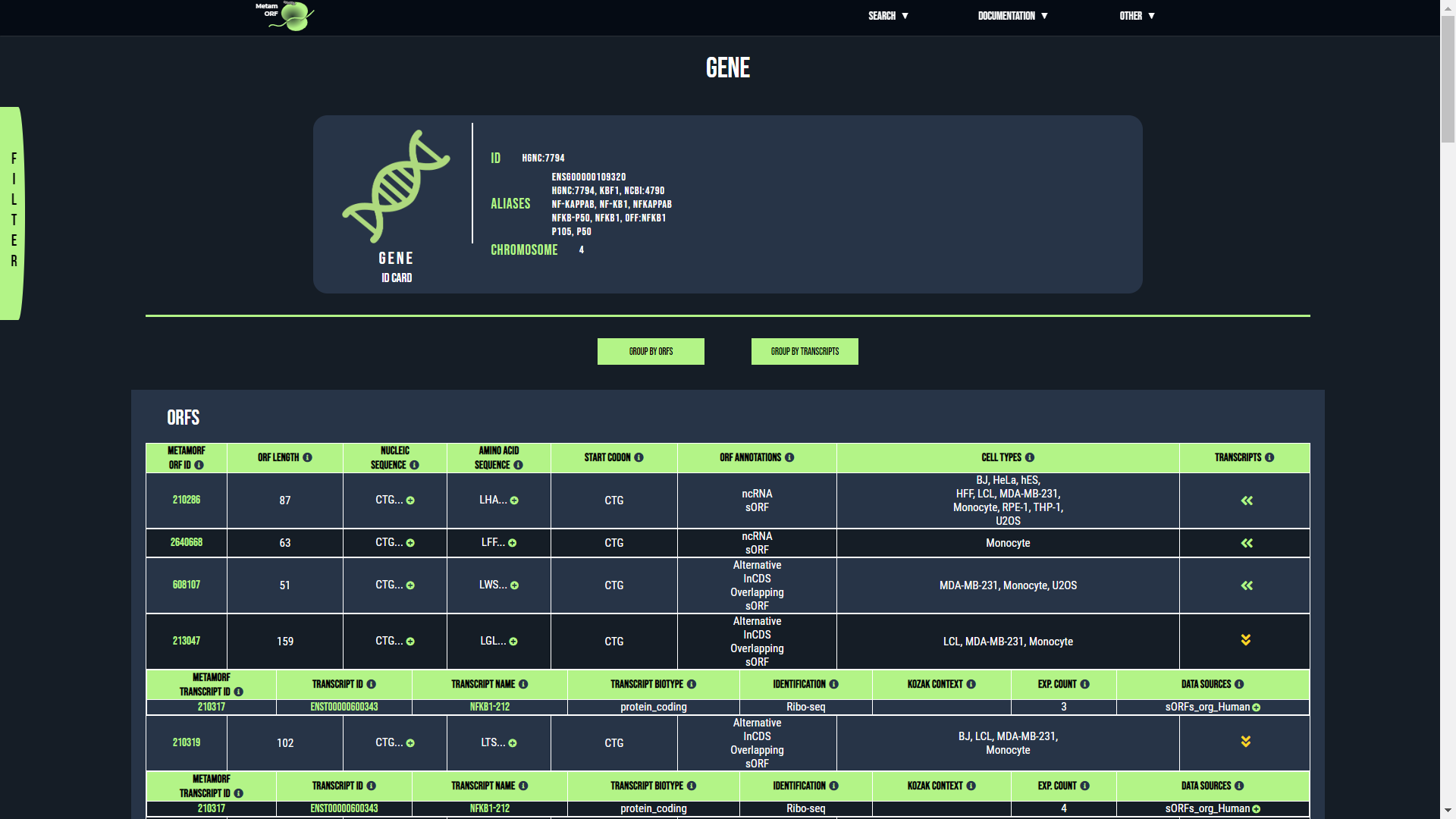
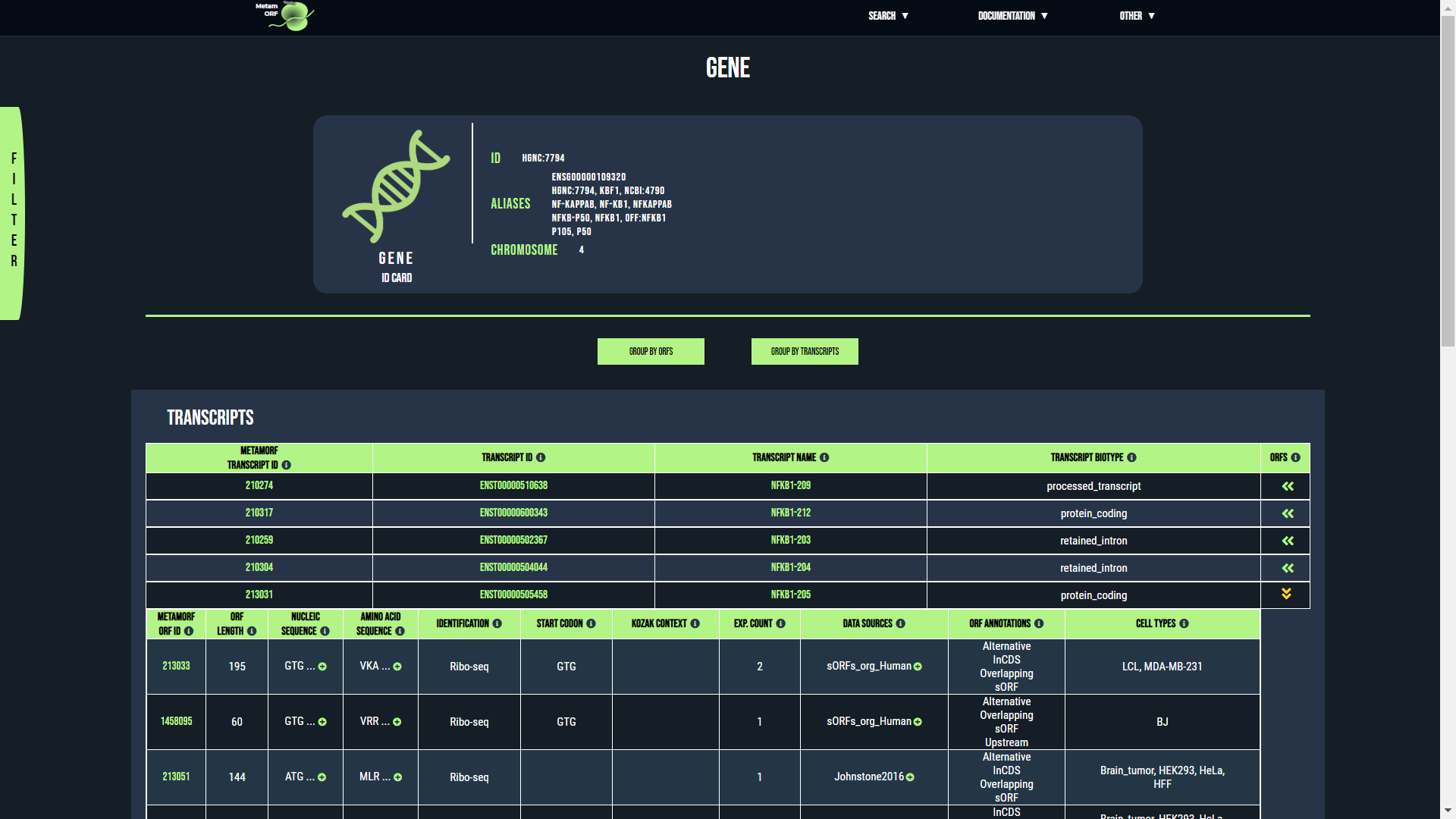
The gene ID card provides the following information:
- ID: The gene ID. Usually the HGNC ID for H. sapiens genes and the NCBI ID for M. musculus genes.
- Aliases: A comma-separated list of symbols, IDs and/or aliases known for the gene.
- Chromosome: The name of the chromosome (or scaffold) the gene is located on.
The rows registering ORFs contain the following columns:
- MetamORF ORF ID: The MetamORF ID of the ORF. This is an unique ID referring to the ORF. You may click on it to access to the ORF centric view corresponding to the entry.
- ORF length: The genomic length of the ORF (in bp). This length is defined as the sum of the lengths of each exon constituting the ORF (thus excluding its eventual introns) and includes both the start and the stop codons.
- Nucleic sequence: The nucleic sequence of the ORF. Only the three first nucleotids are displayed, click on the button to display the full sequence in a pop-up or on the button to display the full sequence in a fasta-like format in a new window.
- Amino acid sequence: The amino acid sequence of the ORF. Only the three first amino acids are displayed, click on the button to display the full sequence in a pop-up or on the button to display the full sequence in a fasta-like format in a new window.
- Identification: The method of identification used to identify the ORF. MetamORF currently integrates data from three main type of identification methods: bioinformatic predictions, ribosome profiling experiments and mass spectrometry experiments (either proteomics or proteogenomics). See the data sources section of the advances documentation for more information about this. As this value is dependent of both the ORF and the transcript, it is provided in the ORF table when results are grouped by transcripts and in the transcript table when results are grouped by ORFs.
- Start codon: The start codon sequence of the ORF.
- Kozak context: The Kozak context computed by our algorithm for the ORF on the transcript. See the Kozak contexts section of the advanced documentation for more details regarding the nomenclature we use. As this value is dependent of both the ORF and the transcript, it is provided in the ORF table when results are grouped by transcripts and in the transcript table when results are grouped by ORFs.
- Exp. count: The number of original datasets that identifed the ORF on the transcript. As this value is dependent of both the ORF and the transcript, it is provided in the ORF table when results are grouped by transcripts and in the transcript table when results are grouped by ORFs.
- Data sources: The data sources in which the ORF has been identified. Click on the button to display all the original IDs in a pop-up. See the data sources section of the advanced documentation for more details regarding the information related to the data sources and the original ORF IDs. As this value is dependent of both the ORF and the transcript, it is provided in the ORF table when results are grouped by transcripts and in the transcript table when results are grouped by ORFs.
- ORF annotations: A comma-separated list of the annotations computed by our algorithm for the ORF. See the section dedicated to ORF annotations in the advanced documentation for more details regarding the nomenclature we use. When the results are grouped by ORFs, all the annotations computed for the ORF are displayed (i.e. all the transcripts on which the ORF has already been reported are considered). When the results are grouped by transcripts, only the annotations computed for the ORF on this particular transcript are displayed.
- Cell types: A comma-separated list of the cell types in which the ORF has already been identified.. When the results are grouped by ORFs, all the cell types in which the ORF has already been described are displayed (i.e. all the transcripts on which the ORF has already been reported are considered). When the results are grouped by transcripts, only the cell types in which the ORF has been described on this particular transcript are displayed.
- Transcripts: When the results are grouped by ORFs, click on the button to display all the transcripts on which the ORF has already been reported.
The rows registering transcripts contain the following columns:
- MetamORF transcript ID: The MetamORF ID of the transcript. This is an arbitrary ID that does not correspond to any official (Ensembl, NCBI...) transcript ID or external reference. You may click on it to access to the transcript centric view corresponding to the entry.
- Transcript ID: The official transcript ID (usually an Ensembl ID, e.g. ENST00000395565). You may click on it to access to the transcript centric view corresponding to the entry.
- Transcript name: The transcript name (e.g. MDK-202). You may click on it to access to the transcript centric view corresponding to the entry.
- Transcript biotype: The biotype of the transcript (as defined by Ensembl).
- ORFs: When the results are grouped by transcripts, click on the button to display all the ORFs that have been reported as harborded by the transcript.
Transcript page
From this page, you will be able to get information regarding the ORFs which are related a particular transcript. The first section describes the transcript itself, the second its gene and the third the ORFs it harbors.
Example of transcript page
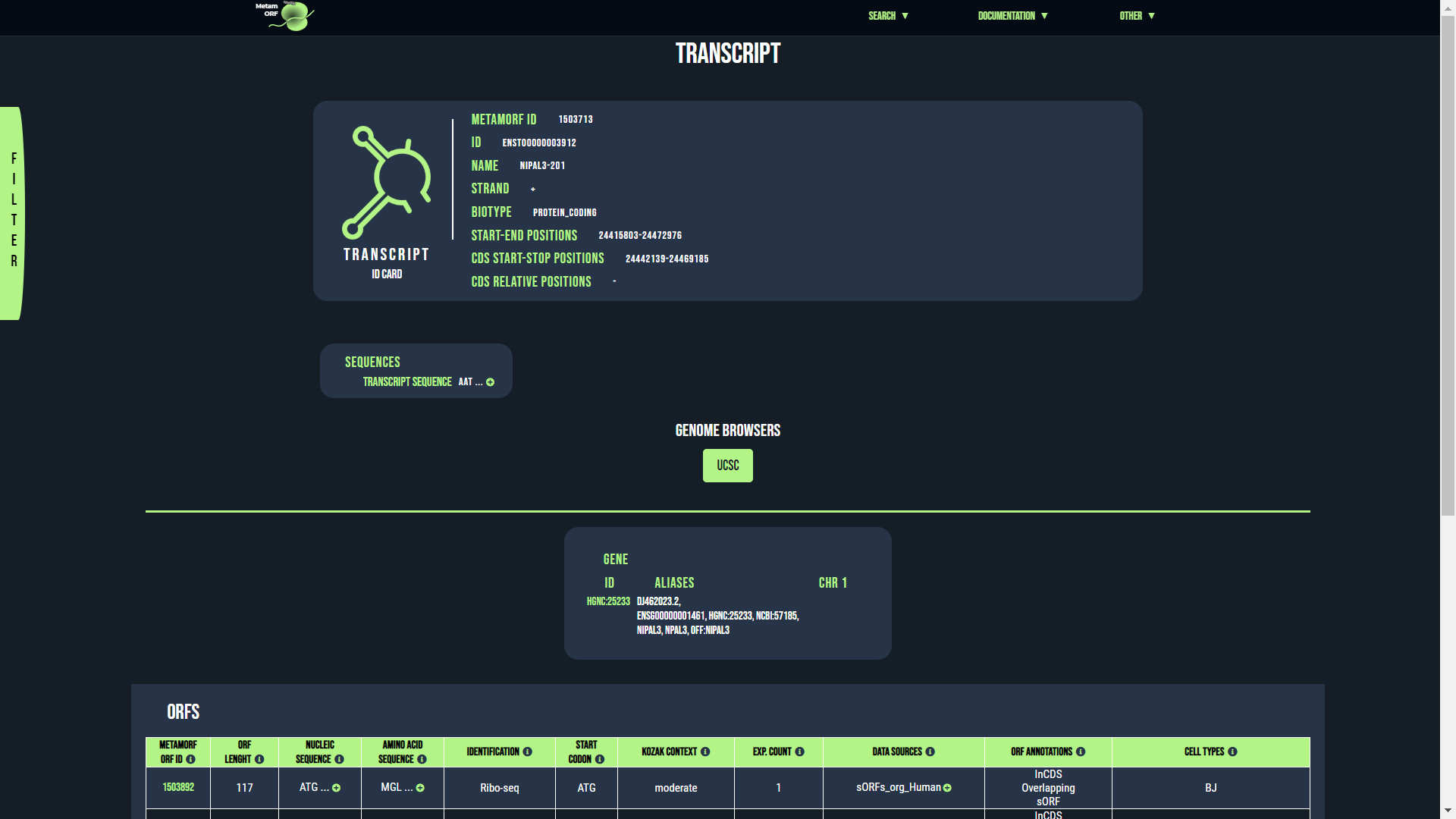
The transcript ID card is made of two blocks which provide the following information:
- MetamORF ID: The MetamORF ID of the transcript. This is an arbitrary ID that does not correspond to any official (Ensembl, NCBI...) transcript ID or external reference.
- ID: The official transcript ID (usually an Ensembl ID, e.g. ENST00000395565).
- Name: The transcript name (e.g. MDK-202).
- Strand: The strand of the transcript (+ or -)
- Biotype: The biotype of the transcript (as defined by Ensembl).
- Start-end positions: The absolute genomic positions (i.e. the positions on the chromosome) of the start and end of the transcript (in a 1-based system).
- CDS start-stop positions: The absolute genomic positions (i.e. the positions on the chromosome) of the start and stop codons of the main (canonical) coding sequence of the transcript (i.e. the CDS, in a 1-based system). These values may only exist for coding transcripts.
- CDS relative positions: The relative positions of the start and stop codons of the canonical coding sequence (CDS) of the transcript. These values may only exist for coding transcripts.
- Transcript sequence: The sequence of the transcript (including its 5' and 3' UTRs).
The gene ID card provides the following information:
- ID: The gene ID. Usually the HGNC ID for H. sapiens genes and the NCBI ID for M. musculus genes. You may click on it to access to the gene centric view corresponding to the entry.
- Alias: A comma-separated list of symbols, IDs and/or aliases known for the gene.
- Chr: The name of the chromosome (or scaffold) the gene is located on.
The rows registering ORFs contain the following columns:
- MetamORF ORF ID: The MetamORF ID of the ORF. This is an unique ID referring to the ORF. You may click on it to access to the ORF centric view corresponding to the entry.
- ORF length: The genomic length of the ORF (in bp). This length is defined as the sum of the lengths of each exon constituting the ORF (thus excluding its eventual introns) and includes both the start and the stop codons.
- Nucleic sequence: The nucleic sequence of the ORF. Only the three first nucleotids are displayed, click on the button to display the full sequence in a pop-up or on the button to display the full sequence in a fasta-like format in a new window.
- Amino acid sequence: The amino acid sequence of the ORF. Only the three first amino acids are displayed, click on the button to display the full sequence in a pop-up or on the button to display the full sequence in a fasta-like format in a new window.
- Identification: The method of identification used to identify the ORF. MetamORF currently integrates data from three main type of identification methods: bioinformatic predictions, ribosome profiling experiments and mass spectrometry experiments (either proteomics or proteogenomics). See the data sources section of the advances documentation for more information about this.
- Start codon: The start codon sequence of the ORF.
- Kozak context: The Kozak context computed by our algorithm for the ORF on the transcript. See the Kozak contexts section of the advanced documentation for more details regarding the nomenclature we use.
- Exp. count: The number of original datasets that identifed the ORF on the transcript.
- Data sources: The data sources in which the ORF has been identified. Click on the button to display all the original IDs in a pop-up. See the data sources section of the advanced documentation for more details regarding the information related to the data sources and the original ORF IDs.
- ORF annotations: A comma-separated list of the annotations computed by our algorithm for the ORF on the transcript. See the section dedicated to ORF annotations in the advanced documentation for more details regarding the nomenclature we use.
- Cell types: A comma-separated list of the cell types in which the ORF has already been identified on the transcript.
ORF page
From this view, you will be able to get information regarding the transcripts and the genes which are related to a particular ORF. The first section describes the ORF itself, the second the transcripts and genes related to it.Example of ORF page
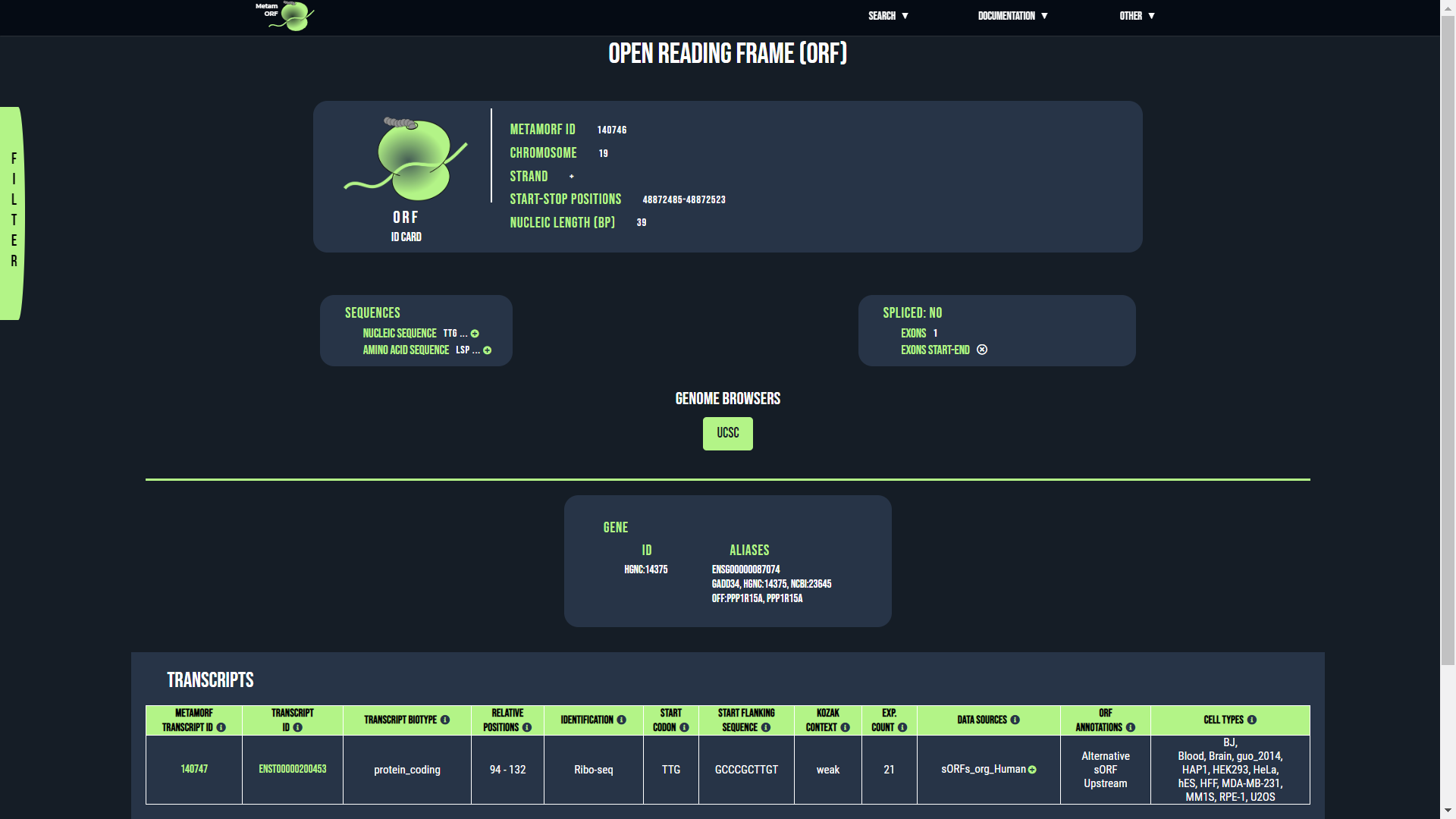
The ORF ID card is made of three blocks which provides the following information:
- MetamORF ID: The MetamORF ID of the ORF. This is an unique ID referring to the ORF.
- Chromosome: The name of the chromosome (or scaffold) the ORF is located on.
- Strand: The strand of the ORF (+ or -)
- Start-stop positions: The absolute genomic positions (i.e. the positions on the chromosome) of the start and stop codons of the ORF (in a 1-based system).
- Nucleic length (bp): The genomic length of the ORF (in bp). This length is defined as the sum of the lengths of each exon constituting the ORF (thus excluding its eventual introns) and includes both the start and the stop codons.
- Nucleic sequence: The nucleic sequence of the ORF. Only the 35 first nucleotids are displayed, click on the button to display the full sequence in a pop-up or on the button to display the full sequence in a fasta-like format in a new window.
- Amino acid sequence: The amino acid sequence of the ORF. Only the 35 first amino acids are displayed, click on the button to display the full sequence in a pop-up or on the button to display the full sequence in a fasta-like format in a new window.
- Spliced: Is the ORF made of several exons?
- Exons: The number of exons constituting the ORF
- Exons start-end: The list of exon start and end positions. Caution: these positions are provided in the order they appear on the transcript. The exons are provided in the order on the transcript, i.e. the first set of position corresponds to the positions of the first exon, the second set to the positions of the second exon and so on. In addition, for a set of position, the first value always corresponds to the position of the first nucleotide of the first codon of the exon whilst the second value always correspond to the position of the third nucleotide of the last codon of the exon. Hence, contrary to the ORF and transcript coordinates, there is no need to reverse the positions when the ORF is located on the minus strand.
When the ORF is associated to one single gene, the gene ID card provides the following information:
- ID: The gene ID. Usually the HGNC ID for H. sapiens genes and the NCBI ID for M. musculus genes. You may click on it to access to the gene centric view corresponding to the entry.
- Alias: A comma-separated list of symbols, IDs and/or aliases known for the gene.
The rows registering transcripts contain the following columns:
- MetamORF transcript ID: The MetamORF ID of the transcript. This is an arbitrary ID that does not correspond to any official (Ensembl, NCBI...) transcript ID or external reference. You may click on it to access to the transcript centric view corresponding to the entry.
- Transcript ID: The official transcript ID (usually an Ensembl ID, e.g. ENST00000395565). You may click on it to access to the transcript centric view corresponding to the entry.
- Transcript biotype: The biotype of the transcript (as defined by Ensembl).
- Gene ID: The gene ID. Usually the HGNC ID for H. sapiens genes and the NCBI ID for M. musculus genes. You may click on it to access to the gene centric view corresponding to the entry. This columns does not appear (and is replaced by the Gene ID card) when the ORF is associated with one single gene.
- Relative positions: The relative positions of the start and stop codons of the ORF on the transcript.
- Identification: The method of identification used to identify the ORF. MetamORF currently integrates data from three main type of identification methods: bioinformatic predictions, ribosome profiling experiments and mass spectrometry experiments (either proteomics or proteogenomics). See the data sources section of the advances documentation for more information about this.
- Start codon: The start codon sequence of the ORF.
- Start flanking sequence: The sequence flanking the start codon of the ORF on the transcript. This sequence registered the nucleotides from -6 to +4 positions, where +1 corresponds to the first nucleotide of the ORF start codon.
- Kozak context: The Kozak context computed by our algorithm for the ORF on the transcript. See the Kozak contexts section of the advanced documentation for more details regarding the nomenclature we use.
- Exp. count: The number of original datasets that identifed the ORF on the transcript.
- Data sources: The data sources in which the ORF has been identified. Click on the button to display all the original IDs in a pop-up. See the data sources section of the advanced documentation for more details regarding the information related to the data sources and the original ORF IDs.
- ORF annotations: A comma-separated list of the annotations computed by our algorithm for the ORF on the transcript. See the section dedicated to ORF annotations in the advanced documentation for more details regarding the nomenclature we use.
- Cell types:A comma-separated list of the cell types in which the ORF has already been identified on the transcript.
Locus page
From this page, you will be able to get information regarding the ORFs located on a particular locus or in a particular area of the genome.
Example of locus page
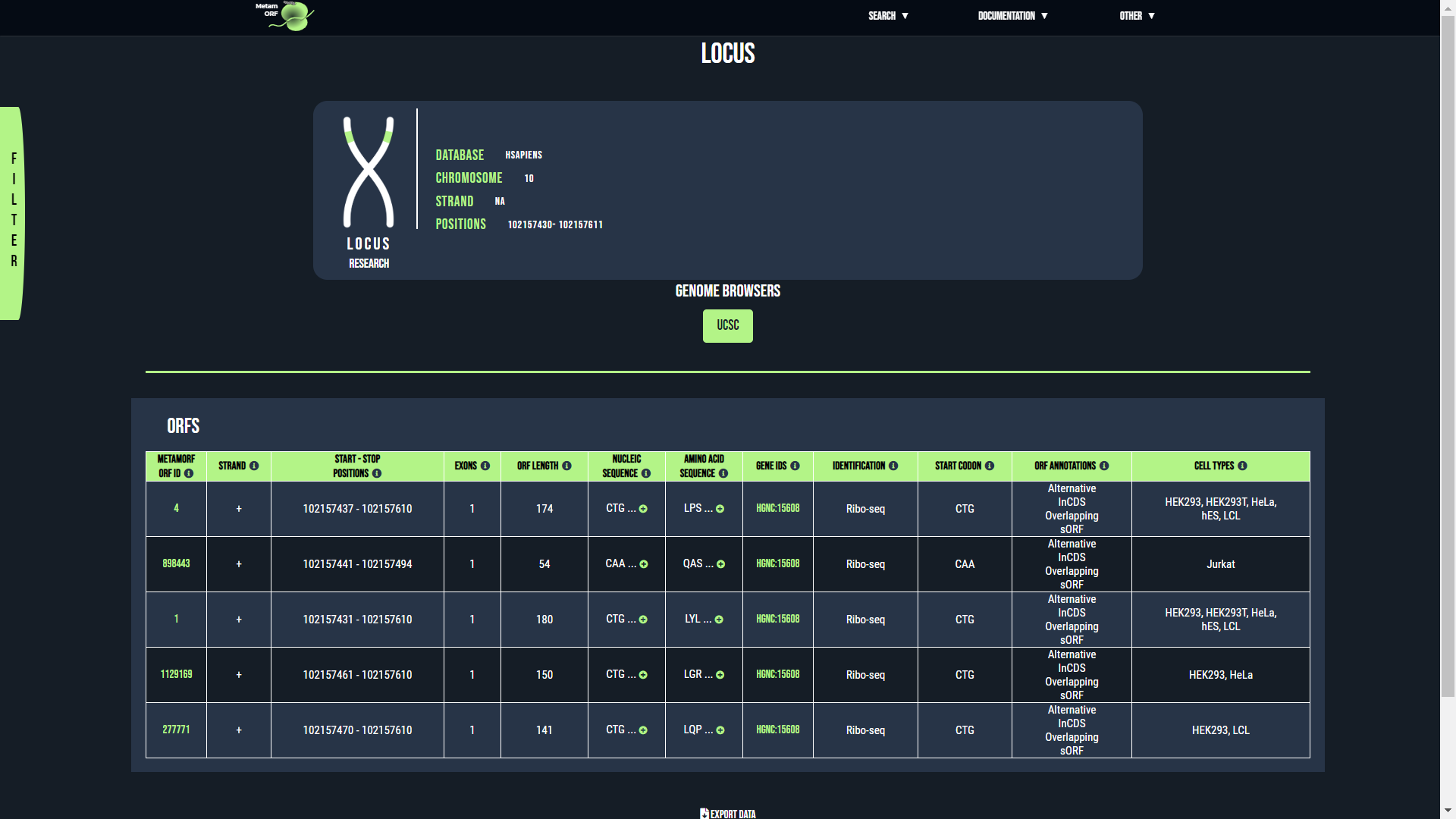
The locus card provides the following information:
- Database: The name of the database queried (hsapiens or mmusculus).
- Chromosome: The chromosome name provided.
- Strand: The strand (+ or -, replaced by 'NA' when not provided).
- Positions: The lower and upper bound of the area considered.
The rows registering ORFs contain the following columns:
- MetamORF ORF ID: The MetamORF ID of the ORF. This is an unique ID referring to the ORF. You may click on it to access to the ORF centric view corresponding to the entry.
- Strand: The strand of the ORF (+ or -)
- Start - stop positions: The absolute genomic positions (i.e. the positions on the chromosome) of the start and stop codons of the ORF (in a 1-based system).
- Exons: The number of exons constituting the ORF
- ORF length: The genomic length of the ORF (in bp). This length is defined as the sum of the lengths of each exon constituting the ORF (thus excluding its eventual introns) and includes both the start and the stop codons.
- Nucleic sequence: The nucleic sequence of the ORF. Only the three first nucleotids are displayed, click on the button to display the full sequence in a pop-up or on the button to display the full sequence in a fasta-like format in a new window.
- Amino acid sequence: The amino acid sequence of the ORF. Only the three first amino acids are displayed, click on the button to display the full sequence in a pop-up or on the button to display the full sequence in a fasta-like format in a new window.
- Gene IDs: The gene ID. Usually the HGNC ID for H. sapiens genes and the NCBI ID for M. musculus genes.
- Identification: The method of identification used to identify the ORF. MetamORF currently integrates data from three main type of identification methods: bioinformatic predictions, ribosome profiling experiments and mass spectrometry experiments (either proteomics or proteogenomics). See the data sources section of the advances documentation for more information about this.
- Start codon: The start codon sequence of the ORF.
- ORF annotations: A comma-separated list of all the annotations computed by our algorithm for the ORF. This list includes the annotations computed for the ORF for all transcripts. See the section dedicated to ORF annotations in the advanced documentation for more details regarding the nomenclature we use.
- Cell types: A comma-separated list of all the cell types in which the ORF has already been identified.
Filtering the results
On each view, the results can be filtered opening the menu located on the left side of the page.
Filtering the results

Several types of filters are common to all four pages (gene, transcript, ORF and locus pages) whilst some other may be specific to some pages. These filters are described below.
When several filters of the same type (e.g. Cell type HEK293 and Cell type HeLa) are selected at the same time, all the entries matching at least one of the condition will be selected (the operation performed is equivalent to an union).
When several type of filters (e.g. Cell type HEK293 and ORF annotation Upstream) are selected at the same time, only entries matching all the criteria will be selected (the operation performed is equivalent to an intersection).
- Identification method: This filter allows to select exclusively the ORFs that were detected with the identification method(s) selected with the filter. See the the advanced documentation section dedicated to the data sources for more information about the methods registered. If several identification methods are selected, all entries matching at least one of the criteria will be displayed.
-
Start codon: This filter allows to select the ORFs with a particular start codon. Three nucleotides must be provided respecting the standard IUB/IUPAC nucleic acid codes (A, T, G, C, N allowed). Lower-case letters are accepted and are mapped into upper-case. The following wildcards may replace one or several of the nucleotides:
- N for any base (A/T/G/C)
- W for weak (A/T)
- S for strong (G/C)
- R for purines (G/A)
- Y for pyrimidine (T/C)
- K for keto (G,T)
- M for amino (A/C)
- B for G/T/C
- D for G/A/T
- H for A/C/T
- V for G/C/A
- Kozak context: This filter allows to select the ORFs located on transcripts where their Kozak context matches the one selected. the advanced documentation section dedicated to the data sources for more information about the Kozak contexts definitions. If several contexts are selected, all entries matching at least one of the criteria will be displayed.
- ORF length: This filter allows to select the ORFs within a specific range of size. A minimal or a maximal size or both of them can be provided to filter the results. The filter uses the Genomic lengths of the ORFs, defined as the sum of the lengths (in bp) of each exon constituting the ORF (thus excluding its eventual introns) and including both the start and the stop codons. The ORFs that equals the minimal and / or the maximal value provided will be selected.
- Transcript biotype: The filters of this section allow to select the ORFs located on one (or several) specific transcript biotypes. Biotype definitions may be found on the Ensembl website. If several biotypes are selected, all entries matching at least one of the criteria will be displayed.
- ORF annotations: The filters of this section allow to select the ORFs annotated in a particular category (short, upstream...). Four categories of annotations are defined depending on the criteria used by our algorithm to perform the annotation: length, biotype, relative position and reading frame. Please see the section of the advanced documentation dedicated to the ORF annotations for more details regarding the nomenclature we use. If several annotations of the same category are selected (e.g. Upstream and Overlaping), all entries matching at least one of the criteria will be displayed. If several annotations of different categories are selected (e.g. Upstream and sORF), all entries matching all the criteria will be displayed.
- Cell types: The filters of this section allow to select the ORFs that have been identified in some particular cell types. Here, a cell type can actually be a cell line, a tissue, an organ or eventually a sample from tissue or organ under pathological condition, depending on the information provided in the original data sources. If several cell types are selected, all entries matching at least one of the criteria will be displayed.
To confirm your selection click on the Filter button. The Restart button allows to reset all the changes that have been recently applied on the filters (i.e. since the last load of the page). The Clear button will delete all the filter selected. When one of these button is used, it is then necessary to refresh the page using the Filter button to get the appropriate results. When a page is loaded for the first time, there are obviously no filters applied.
Exporting the results
The four pages allow to export results of your researches at CSV, FASTA and BED formats. These formats and the data they contain are detailed in this section. Please note that the preparation of the file to download may take some time if you are displaying a large set of data on the browser.
Exporting the results

CSV format
Files exported at the comma-separated values format may be opened with any common spreadsheet software or text editors. CSV files aims to be parsable and are known to contain redundant information.
CSV export from the browser
CSV export from the browser produces a file that starts with a variable number of lines of comments (defined by a #) that register the filters you selected.
The next line contains the columns' headers and is followed by a variable number of lines describing the ORFs. Each row contains an unique ORF. These rows register the following information:
- MetamORF_orf_id: The MetamORF ID of the ORF. This is an unique ID referring to the ORF.
- chromosome: The name of the chromosome (or scaffold) the ORF is located on.
- strand: The strand of the ORF (+ or -)
- start_pos: The absolute genomic positions (i.e. the positions on the chromosome) of the start of the ORF (start codon for the ORFs located on the forward strand, stop codon for the ORFs located on the reverse strand, in a 1-based system).
- stop_pos: The absolute genomic positions (i.e. the positions on the chromosome) of the stop of the ORF (stop codon for the ORFs located on the forward strand, start codon for the ORFs located on the reverse strand, in a 1-based system).
- exons: An underscore-separated list of all exon start positions. Caution: these positions are provided in the order they appear on the transcript. Hence, the first value always corresponds to the position of the first nucleotide of the first codon of the first exon, the second value corresponds to the position of the first nucleotide of the first codon of the second exon, and so on.
- exon_starts: An underscore-separated list of all exon start positions. Caution: these positions are provided in the order they appear on the transcript. Hence, the first value always corresponds to the position of the first nucleotide of the first codon of the first exon, the second value corresponds to the position of the first nucleotide of the first codon of the second exon, and so on.
- exon_ends: An underscore-separated list of all exon end positions. Caution: these positions are provided in the order they appear on the transcript. Hence, the first value always corresponds to the position of the last nucleotide of the last codon of the first exon, the second value corresponds to the position of the last nucleotide of the last codon of the second exon, and so on.
- orf_length: The genomic length of the ORF (in bp). This length is defined as the sum of the lengths of each exon constituting the ORF (thus excluding its eventual introns) and includes both the start and the stop codons.
- sequence_nt: The nucleic sequence of the ORF.
- sequence_aa: The amino acid sequence of the ORF.
CSV export from a gene page
CSV export from gene page produces a file that starts with 3 lines of comments (defined by a #) that register the following information:
- #gene_ID: The gene ID. Usually the HGNC ID for H. sapiens genes and the NCBI ID for M. musculus genes.
- #aliases: A comma-separated list of symbols, IDs and/or aliases known for the gene.
- #chromosome: The name of the chromosome (or scaffold) the gene is located on.
The fourth line contains the columns' headers and is followed by a variable number of lines describing the ORFs and transcripts related to the gene. Each row contains an unique ORF - transcript association. These rows register the following information:
- MetamORF_orf_id: The MetamORF ID of the ORF. This is an unique ID referring to the ORF.
- chromosome: The name of the chromosome (or scaffold) the ORF is located on.
- strand: The strand of the ORF (+ or -)
- start_pos: The absolute genomic positions (i.e. the positions on the chromosome) of the start of the ORF (start codon for the ORFs located on the forward strand, stop codon for the ORFs located on the reverse strand, in a 1-based system).
- stop_pos: The absolute genomic positions (i.e. the positions on the chromosome) of the stop of the ORF (stop codon for the ORFs located on the forward strand, start codon for the ORFs located on the reverse strand, in a 1-based system).
- exons: An underscore-separated list of all exon start positions. Caution: these positions are provided in the order they appear on the transcript. Hence, the first value always corresponds to the position of the first nucleotide of the first codon of the first exon, the second value corresponds to the position of the first nucleotide of the first codon of the second exon, and so on.
- exon_starts: An underscore-separated list of all exon start positions. Caution: these positions are provided in the order they appear on the transcript. Hence, the first value always corresponds to the position of the first nucleotide of the first codon of the first exon, the second value corresponds to the position of the first nucleotide of the first codon of the second exon, and so on.
- exon_ends: An underscore-separated list of all exon end positions. Caution: these positions are provided in the order they appear on the transcript. Hence, the first value always corresponds to the position of the last nucleotide of the last codon of the first exon, the second value corresponds to the position of the last nucleotide of the last codon of the second exon, and so on.
- orf_length: The genomic length of the ORF (in bp). This length is defined as the sum of the lengths of each exon constituting the ORF (thus excluding its eventual introns) and includes both the start and the stop codons.
- sequence_nt: The nucleic sequence of the ORF.
- sequence_aa: The amino acid sequence of the ORF.
- MetamORF_transcript_id: The MetamORF ID of the transcript. This is an arbitrary ID that does not correspond to any official (Ensembl, NCBI...) transcript ID or external reference.
- transcript_id: The official transcript ID (usually an Ensembl ID, e.g. ENST00000395565).
- transcript_name: The transcript name (e.g. MDK-202).
- rna_biotype: The biotype of the transcript (as defined by Ensembl).
- rel_start_pos: The relative position of the start of the ORF on the transcript (start codon for the ORFs located on the forward strand, stop codon for the ORFs located on the reverse strand, in a 1-based system).
- rel_stop_pos: The relative position of the stop of the ORF on the transcript (stop codon for the ORFs located on the forward strand, start codon for the ORFs located on the reverse strand, in a 1-based system).
- predicted: Has the ORF already been identified by bioinformatic prediction?
- ribo_seq: Has the ORF already been identified by ribosome profiling?
- ms: Has the ORF already been identified by mass spectrometry (proteomics, peptidomics or proteogenomics)?
- start_codon_seq: The start codon sequence of the ORF.
- start_flanking_seq: The sequence flanking the start codon of the ORF on the transcript. This sequence registered the nucleotides from -6 to +4 positions, where +1 corresponds to the first nucleotide of the ORF start codon.
- kozak_context: The Kozak context computed by our algorithm for the ORF on the transcript. See the Kozak contexts section of the advanced documentation for more details regarding the nomenclature we use.
- exp_count: The number of original datasets that identifed the ORF on the transcript.
- orf_annotations: A comma-separated list of the annotations computed by our algorithm for the ORF on the transcript. See the section dedicated to ORF annotations in the advanced documentation for more details regarding the nomenclature we use.
- cell_types: A comma-separated list of the cell types in which the ORF has already been identified on the transcript.
CSV export from a transcript page
CSV export from transcript page produces a file that starts with 9 lines of comments (defined by a #) that register the following information:
- #MetamORF_transcript_id: The MetamORF ID of the transcript. This is an arbitrary ID that does not correspond to any official (Ensembl, NCBI...) transcript ID or external reference.
- #ID: The official transcript ID (usually an Ensembl ID, e.g. ENST00000395565).
- #name: The transcript name (e.g. MDK-202).
- #position: The transcript absolutes coordinates at the format chrNameStrand:start-end, where chrName is the chromosome (or scaffold) name, preceed by the 'chr' prefix, Strand is the transcript strand (+ or -), start is the start of the transcript (i.e. the coordinates of the first nucleotide for the transcript located on the forward strand, the coordinates of the last nucleotide for the transcripts located on the reverse strand) and end is the end of the transcript (i.e. the coordinates of the last nucleotide for the transcript located on the forward strand, the coordinates of the first nucleotide for the transcripts located on the reverse strand) (in a 1-based coordinates system).
- #biotype: The biotype of the transcript (as defined by Ensembl).
- #CDS_positions: The absolute genomic positions (i.e. the positions on the chromosome) of the start and stop codons of the main (canonical) coding sequence of the transcript (i.e. the CDS, in a 1-based system). These values may only exist for coding transcripts.
- #CDS_rel_positions: The relative positions of the start and stop codons of the canonical coding sequence (CDS) of the transcript. These values may only exist for coding transcripts.
- #gene_id: The gene ID. Usually the HGNC ID for H. sapiens genes and the NCBI ID for M. musculus genes.
- #sequence: The sequence of the transcript (including its 5' and 3' UTRs).
The tenth line contains the columns' headers and is followed by a variable number of lines describing the ORFs related to the transcript. Each row contains an unique ORF. These rows contain the following information:
- MetamORF_orf_id: The MetamORF ID of the ORF. This is an unique ID referring to the ORF.
- chromosome: The name of the chromosome (or scaffold) the ORF is located on.
- strand: The strand of the ORF (+ or -)
- start_pos: The absolute genomic positions (i.e. the positions on the chromosome) of the start of the ORF (start codon for the ORFs located on the forward strand, stop codon for the ORFs located on the reverse strand, in a 1-based system).
- stop_pos: The absolute genomic positions (i.e. the positions on the chromosome) of the stop of the ORF (stop codon for the ORFs located on the forward strand, start codon for the ORFs located on the reverse strand, in a 1-based system).
- exons: An underscore-separated list of all exon start positions. Caution: these positions are provided in the order they appear on the transcript. Hence, the first value always corresponds to the position of the first nucleotide of the first codon of the first exon, the second value corresponds to the position of the first nucleotide of the first codon of the second exon, and so on.
- exon_starts: An underscore-separated list of all exon start positions. Caution: these positions are provided in the order they appear on the transcript. Hence, the first value always corresponds to the position of the first nucleotide of the first codon of the first exon, the second value corresponds to the position of the first nucleotide of the first codon of the second exon, and so on.
- exon_ends: An underscore-separated list of all exon end positions. Caution: these positions are provided in the order they appear on the transcript. Hence, the first value always corresponds to the position of the last nucleotide of the last codon of the first exon, the second value corresponds to the position of the last nucleotide of the last codon of the second exon, and so on.
- orf_length: The genomic length of the ORF (in bp). This length is defined as the sum of the lengths of each exon constituting the ORF (thus excluding its eventual introns) and includes both the start and the stop codons.
- sequence_nt: The nucleic sequence of the ORF.
- sequence_aa: The amino acid sequence of the ORF.
- rel_start_pos: The relative position of the start of the ORF on the transcript (start codon for the ORFs located on the forward strand, stop codon for the ORFs located on the reverse strand, in a 1-based system).
- rel_stop_pos: The relative position of the stop of the ORF on the transcript (stop codon for the ORFs located on the forward strand, start codon for the ORFs located on the reverse strand, in a 1-based system).
- predicted: Has the ORF already been identified by bioinformatic prediction?
- ribo_seq: Has the ORF already been identified by ribosome profiling?
- ms: Has the ORF already been identified by mass spectrometry (proteomics, peptidomics or proteogenomics)?
- start_codon_seq: The start codon sequence of the ORF.
- start_flanking_seq: The sequence flanking the start codon of the ORF on the transcript. This sequence registered the nucleotides from -6 to +4 positions, where +1 corresponds to the first nucleotide of the ORF start codon.
- kozak_context: The Kozak context computed by our algorithm for the ORF on the transcript. See the Kozak contexts section of the advanced documentation for more details regarding the nomenclature we use.
- exp_count: The number of original datasets that identifed the ORF on the transcript.
- orf_annotations: A comma-separated list of the annotations computed by our algorithm for the ORF on the transcript. See the section dedicated to ORF annotations in the advanced documentation for more details regarding the nomenclature we use.
- cell_types: A comma-separated list of the cell types in which the ORF has already been identified on the transcript.
CSV export from an ORF page
CSV export from ORF page produces a file that starts with 7 lines of comments (defined by a #) that register the following information:
- #MetamORF_orf_id: The MetamORF ID of the ORF. This is an unique ID referring to the ORF.
- #positions: The ORF absolutes coordinates at the format chrNameStrand:start-stop, where chrName is the chromosome (or scaffold) name, preceed by the 'chr' prefix, Strand is the ORF strand (+ or -), start is the start of the ORF (i.e. the coordinates of the start codon for the ORFs located on the forward strand, the coordinates of the stop codon for the ORFs located on the reverse strand) and stop is the stop of the ORF (i.e. the coordinates of the stop codon for the ORFs located on the forward strand, the coordinates of the start codon for the ORFs located on the reverse strand) (in a 1-based coordinates system).
- #exons: The number of exons constituting the ORF
- #exons_pos:
- #orf_length: The genomic length of the ORF (in bp). This length is defined as the sum of the lengths of each exon constituting the ORF (thus excluding its eventual introns) and includes both the start and the stop codons.
- #sequence_nt: The nucleic sequence of the ORF.
- #sequence_aa: The amino acid sequence of the ORF.
The eigth line contains the columns' headers and is followed by a variable number of lines describing the transcripts related to the ORF. Each row contains an unique transcript (with its gene information). These rows contain the following information:
- MetamORF_transcript_id: The MetamORF ID of the transcript. This is an arbitrary ID that does not correspond to any official (Ensembl, NCBI...) transcript ID or external reference.
- transcript_id: The official transcript ID (usually an Ensembl ID, e.g. ENST00000395565).
- transcript_name: The transcript name (e.g. MDK-202).
- rna_biotype: The biotype of the transcript (as defined by Ensembl).
- rel_start_pos: The relative position of the start of the ORF on the transcript (start codon for the ORFs located on the forward strand, stop codon for the ORFs located on the reverse strand, in a 1-based system).
- rel_stop_pos: The relative position of the stop of the ORF on the transcript (stop codon for the ORFs located on the forward strand, start codon for the ORFs located on the reverse strand, in a 1-based system).
- predicted: Has the ORF already been identified by bioinformatic prediction?
- ribo_seq: Has the ORF already been identified by ribosome profiling?
- ms: Has the ORF already been identified by mass spectrometry (proteomics, peptidomics or proteogenomics)?
- start_codon_seq: The start codon sequence of the ORF.
- start_flanking_seq: The sequence flanking the start codon of the ORF on the transcript. This sequence registered the nucleotides from -6 to +4 positions, where +1 corresponds to the first nucleotide of the ORF start codon.
- kozak_context: The Kozak context computed by our algorithm for the ORF on the transcript. See the Kozak contexts section of the advanced documentation for more details regarding the nomenclature we use.
- exp_count: The number of original datasets that identifed the ORF on the transcript.
- orf_annotations: A comma-separated list of the annotations computed by our algorithm for the ORF on the transcript. See the section dedicated to ORF annotations in the advanced documentation for more details regarding the nomenclature we use.
- cell_types: A comma-separated list of the cell types in which the ORF has already been identified on the transcript.
CSV export from a locus page
CSV export from locus page produces a file that starts with 4 lines of comments (defined by a #) that register the following information:
- #chromosome: The chromosome name of the locus.
- #strand: The strand of the locus (+, - or NA).
- #start: The lower bound of the locus.
- #end: The upper bound of the locus.
The fifth line contains the columns' headers and is followed by a variable number of lines describing the ORFs located in the locus. Each row contains an unique ORF - transcript association. These rows contain the following information:
- MetamORF_orf_id: The MetamORF ID of the ORF. This is an unique ID referring to the ORF.
- chromosome: The name of the chromosome (or scaffold) the ORF is located on.
- strand: The strand of the ORF (+ or -)
- start_pos: The absolute genomic positions (i.e. the positions on the chromosome) of the start of the ORF (start codon for the ORFs located on the forward strand, stop codon for the ORFs located on the reverse strand, in a 1-based system).
- stop_pos: The absolute genomic positions (i.e. the positions on the chromosome) of the stop of the ORF (stop codon for the ORFs located on the forward strand, start codon for the ORFs located on the reverse strand, in a 1-based system).
- exons: An underscore-separated list of all exon start positions. Caution: these positions are provided in the order they appear on the transcript. Hence, the first value always corresponds to the position of the first nucleotide of the first codon of the first exon, the second value corresponds to the position of the first nucleotide of the first codon of the second exon, and so on.
- exon_starts: An underscore-separated list of all exon start positions. Caution: these positions are provided in the order they appear on the transcript. Hence, the first value always corresponds to the position of the first nucleotide of the first codon of the first exon, the second value corresponds to the position of the first nucleotide of the first codon of the second exon, and so on.
- exon_starts: An underscore-separated list of all exon end positions. Caution: these positions are provided in the order they appear on the transcript. Hence, the first value always corresponds to the position of the last nucleotide of the last codon of the first exon, the second value corresponds to the position of the last nucleotide of the last codon of the second exon, and so on.
- orf_length: The genomic length of the ORF (in bp). This length is defined as the sum of the lengths of each exon constituting the ORF (thus excluding its eventual introns) and includes both the start and the stop codons.
- sequence_nt: The nucleic sequence of the ORF.
- sequence_aa: The amino acid sequence of the ORF.
- MetamORF_transcript_id: The MetamORF ID of the transcript. This is an arbitrary ID that does not correspond to any official (Ensembl, NCBI...) transcript ID or external reference.
- transcript_id: The official transcript ID (usually an Ensembl ID, e.g. ENST00000395565).
- transcript_name: The transcript name (e.g. MDK-202).
- rna_biotype: The biotype of the transcript (as defined by Ensembl).
- gene_ID: The gene ID. Usually the HGNC ID for H. sapiens genes and the NCBI ID for M. musculus genes.
- rel_start_pos: The relative position of the start of the ORF on the transcript (start codon for the ORFs located on the forward strand, stop codon for the ORFs located on the reverse strand, in a 1-based system).
- rel_stop_pos: The relative position of the stop of the ORF on the transcript (stop codon for the ORFs located on the forward strand, start codon for the ORFs located on the reverse strand, in a 1-based system).
- predicted: Has the ORF already been identified by bioinformatic prediction?
- ribo_seq: Has the ORF already been identified by ribosome profiling?
- ms: Has the ORF already been identified by mass spectrometry (proteomics, peptidomics or proteogenomics)?
- start_codon_seq: The start codon sequence of the ORF.
- start_flanking_seq: The sequence flanking the start codon of the ORF on the transcript. This sequence registered the nucleotides from -6 to +4 positions, where +1 corresponds to the first nucleotide of the ORF start codon.
- kozak_context: The Kozak context computed by our algorithm for the ORF on the transcript. See the Kozak contexts section of the advanced documentation for more details regarding the nomenclature we use.
- exp_count: The number of original datasets that identifed the ORF on the transcript.
- orf_annotations: A comma-separated list of the annotations computed by our algorithm for the ORF on the transcript. See the section dedicated to ORF annotations in the advanced documentation for more details regarding the nomenclature we use.
- cell_types: A comma-separated list of the cell types in which the ORF has already been identified on the transcript.
Fasta format
Results may be exported at FASTA format. The file contains either the nucleic (FASTA NT) or the amino acid (FASTA AA) sequences. If you are not familiar with the fasta file formats, we strongly suggest to refer to the UniProt documentation for more information. Files exported from MetamORF at the FASTA format contains long headers respecting the UniProt conventions.
The headers first contains three pipe ('|')-separated values in the format db|UniqueIdentifier|EntryName:
- db: 'mORF' refers to MetamORF.
- UniqueIdentifier: The unique identifier of the ORF in MetamORF (with the 'ORF' prefix, ORF4)
- EntryName: The unique identifier of the ORF in MetamORF (with the 'ORF' prefix) followed by the species code (underscore-separated, ORF277771_HUMAN))
The following elements of the header are space-separated and provide additional information regarding the ORF OS=OrganismName OX=OrganismIdentifier chr=ChrName strand=Strand start_pos=StartPos stop_pos=StopPos orf_annotations=Annotations cell_types=CellTypes (OS=OrganismName OX=OrganismIdentifier chr=ChrName strand=Strand start_pos=StartPos stop_pos=StopPos for exports performed from the ORF browser):
- OrganismName: The scientific name of the organism (i.e. Homo sapiens or Mus musculus).
- OrganismIdentifier: The unique identifier of the source arganism, assigned by the NCBI (i.e. Homo sapiens or Mus musculus).
- chr: A comma-separated list of the cell types in which the ORF has already been identified on the transcript.
- strand: The strand of the ORF (+ or -)
- start_pos: The absolute genomic positions (i.e. the positions on the chromosome) of the start of the ORF (start codon for the ORFs located on the forward strand, stop codon for the ORFs located on the reverse strand, in a 1-based system).
- stop_pos: The absolute genomic positions (i.e. the positions on the chromosome) of the stop of the ORF (stop codon for the ORFs located on the forward strand, start codon for the ORFs located on the reverse strand, in a 1-based system).
- annotations: A comma-separated list of all the annotations computed by our algorithm for the ORF. This list includes the annotations computed for the ORF for all transcripts. See the section dedicated to ORF annotations in the advanced documentation for more details regarding the nomenclature we use.
- cell_types: A comma-separated list of all the cell types in which the ORF has already been identified.
- transcript_ids: A comma separated list of official transcript IDs related to the ORF.
BED format
Results may finally be exported at BED format. The file contains information related to the ORFs themselves (and no information regarding the genes or the transcripts). BED files may be used with Genome Browsers (such as IGV, the UCSC Genome Browser or the Ensembl Genome Browser), as well as with various softwares. If you are not familiar with the BED file formats, we strongly suggest to refer to the UCSC documentation for more information. Files exported from MetamORF at the BED format are bed12 files (where blocks are corresponding to ORF exons).
BED 12+5
The ORF browser allows to export the data at both BED and BED 12+5 formats. The BED 12+5 file contains the 12 usual columns found in BED files as well as 5 extra-columns that register the following information:
- A comma-separated list of the official IDs of all the transcripts related to the ORFs (e.g. Ensembl transcript IDs).
- A comma-separated list of the biotypes of all the transcripts related to the ORFs. Caution: The order of this list does not necessarily match the order of the list provided by the first column.
- A comma-separated list of all the cell types in which the ORF has already been identified.
- A comma-separated list of all the annotations computed by our algorithm for the ORF. This list includes the annotations computed for the ORF for all transcripts. See the section dedicated to ORF annotations in the advanced documentation for more details regarding the nomenclature we use.
- A comma-separated list of Kozak context computed by our algorithm for the ORFs. This list includes the contexts computed for the ORF for all transcripts. See the Kozak contexts section of the advanced documentation for more details regarding the nomenclature we use. Caution: The order of this list does not necessarily match the order of the list provided by the first column.
Please, be aware that numerous softwares do not accept bed files containing extra columns. In case of doubt, we advice to use the file at BED12 format.
Advanced documentation
Data sources
Data from computational predictions, ribosome profiling experiments and mass spectrometry expriments (peptidomics, proteomics, proteogenomics) have been integrated in the database.
The following data sources have been integrated in the database:
| Name of the data source | doi | Description | url |
|---|---|---|---|
| Erhard2018 | 10.1038/nmeth.4631 | Erhard et al., Nat. Meth., 2018. "Supplementary Table 3: Identified ORFs (Union of all ORFs detected either by PRICE,RP-BP or ORF-RATER, or contained in the annotation (Ensembl V75))". The two first lines of the file have to be manually removed. | https://www.nature.com/articles/nmeth.4631 |
| Johnstone2016 | 10.15252/embj.201592759 | Johnstone et al., EMBO, 2016. "Dataset EV2: Location and translation data for all analyzed transcripts and ORFs in human". | http://emboj.embopress.org/content/35/7/706.long |
| Laumont2016 | 10.1038/ncomms10238 | Laumont et al., Nat. Commun., 2016. "Supplementary Data 2: List of all cryptic MAPs detected in subject 1. Table presenting the genomic and proteomic features of all cryptic MAPs". The two first rows have to be manually removed. | https://www.nature.com/articles/ncomms10238 |
| Mackowiak2015 | 10.1186/s13059-015-0742-x | Mackowiak et al., Genome Biol., 2015. "Additional file 2: Table S1. All sORF information for human". The file header has to be removed manually (45 first rows). | https://genomebiology.biomedcentral.com/articles/10.1186/s13059-015-0742-x |
| Samandi2017 | 10.7554/eLife.27860 | Samandi et al., eLIFE, 2017. "Homo sapiens alternative protein predictions based on RefSeq GRCh38 (hg38) based on assembly GCF_000001405.26. Release date 01/01/2016.". The TSV file has been used. | https://elifesciences.org/articles/27860 |
| sORFs_org_Human | 10.1093/nar/gkx1130 | Olexiouk et al., Nucl. Ac. Res., 2018. H. sapiens database downloaded from sORFs.org using the Biomart Graphic User Interface. The following parameters were used to query the database: "Homo sapiens" > "no filters" > "select all MAIN_ATTRIBUTES" > "results" > "download data". | https://academic.oup.com/nar/article/46/D1/D497/4621340 |
| Name of the data source | doi | Description | url |
|---|---|---|---|
| Johnstone2016 | 10.15252/embj.201592759 | Johnstone et al., EMBO, 2016. "Dataset EV3: Location and translation data for all analyzed transcripts and ORFs in mouse". | http://emboj.embopress.org/content/35/7/706.long |
| Mackowiak2015 | 10.1186/s13059-015-0742-x | Mackowiak et al., Genome Biol., 2015. "Additional file 3: Table S2. All sORF information for mouse". The file header has to be removed manually (45 first rows). | https://genomebiology.biomedcentral.com/articles/10.1186/s13059-015-0742-x |
| Samandi2017 | 10.7554/eLife.27860 | Samandi et al., eLIFE, 2017. "Mus musculus alternative protein predictions based on annotation version GRCm38. Release date 01/01/2016". The TSV file has been used. | https://elifesciences.org/articles/27860 |
| sORFs_org_Mouse | 10.1093/nar/gkx1130 | Olexiouk et al., Nucl. Ac. Res., 2018. M. musculus database downloaded from sORFs.org using the Biomart Graphic User Interface. The following parameters were used to query the database: "Mus musculus" > "no filters" > "select all MAIN_ATTRIBUTES" > "results" > "download data". | https://academic.oup.com/nar/article/46/D1/D497/4621340 |
The following data set from sORFs.org original datasets for H.sapiens (46 datasets): andreev_2015, cenik_2015, eichorn_2014, ingolia_2014, iwasaki_2016, jan_2014, park_2016, park_2017, werner_2015, zur_2016, calviello_2016, elkon_2015, fritsch_2012, ingolia_2012, jakobsson_2017, liu_Hela_2013, loayza_puch_2013, loayza_puch_2016, malecki_2017, mills_2016, niu_2014, rooijers_2013, rubio_2014, sidrauski_2015, tanenbaum_2015, tirosh_2015, wang_2015, xu_2016, wiita_2013, liu_HEK_2013, gawron_2016, rutkowski_2015, gonzalez_2014, guo_2014, yoon_2014, crappe_2014, zhang_2017, stern_ginossar_2012, stumpf_2013, ji_BJ_2015, lee_2012, su_2015, shi_2017, ji_breast_2015, wein_2014, grow_2015.
The following data set from sORFs.org original datasets for M.musculus (27 datasets): blanco_2016, cho_2015, castaneda_2014, eichorn_bcell_2014, laguesse_2015, deklerck_2015, gerashchenko_2016, hurt_2013, ingolia_2014_mmu, katz_2014, reid_2014, reid_er_2016, thoreen_2012, eichorn_liver_2014, gonzalez_2014_mmu, guo_2010_mmu, diaz_munoz_2015, eichorn_3t3_2014, janich_2015, you_2015, reid_cytosol_2016, jovanovic_2015, gao_mef_2014, fields_2015, ingolia_2011, gao_liver_2014, lee_2012_mmu.
The original IDs may be used to access the entries in the original datasets / databases. As some sources were not providing any ORF IDs, you may find here the IDs we used as "original IDs" for each data source:
- Erhard2018: The "original" IDs look like index_chr:start-stop:strand, where index is the line number of the entry in the original file, chr is the chromosome name (with the 'chr' prefix), start, stop and strand are respectively the ORF start and stop location and the ORF strand in the original annotation version.
- Johnstone2016: The original ID used is the one registered in the column orfID of the data source.
- Laumont2016: The "original" IDs look like index_chr:start-stop:strand, where index is the line number of the entry in the original file, chr is the chromosome name (with the 'chr' prefix), start, stop and strand are respectively the ORF start and stop location and the ORF strand in the original annotation version.
- Mackowiak2015: The original ID used is the one registered in the column orfID of the data source.
- Samandi2017: The original ID used is the one registered in the column Protein accession of the data source.
- sORFs_org_Human: The original ID used is the one registered in the column Sorf ID of the data source and corresponds to the unique ID used on sORFs.org.
- sORFs_org_Mouse: The original ID used is the one registered in the column sORF ID of the data source and corresponds to the unique ID used on sORFs.org.
ORF annotations
Despite the use of a common nomenclature by the majority of the data sources to describe the ORFs, no clear consensus regarding the definitions of these categories or their names has been defined so far. Hence, in order to get common definitions of ORF annotations for all the entries, the ORF annotations has been defined according to five main classes of criteria: the ORF length, the RNA biotype, the position of the ORF on the transcript relatively to the canonical CDS, the reading frame and the ORF strand.
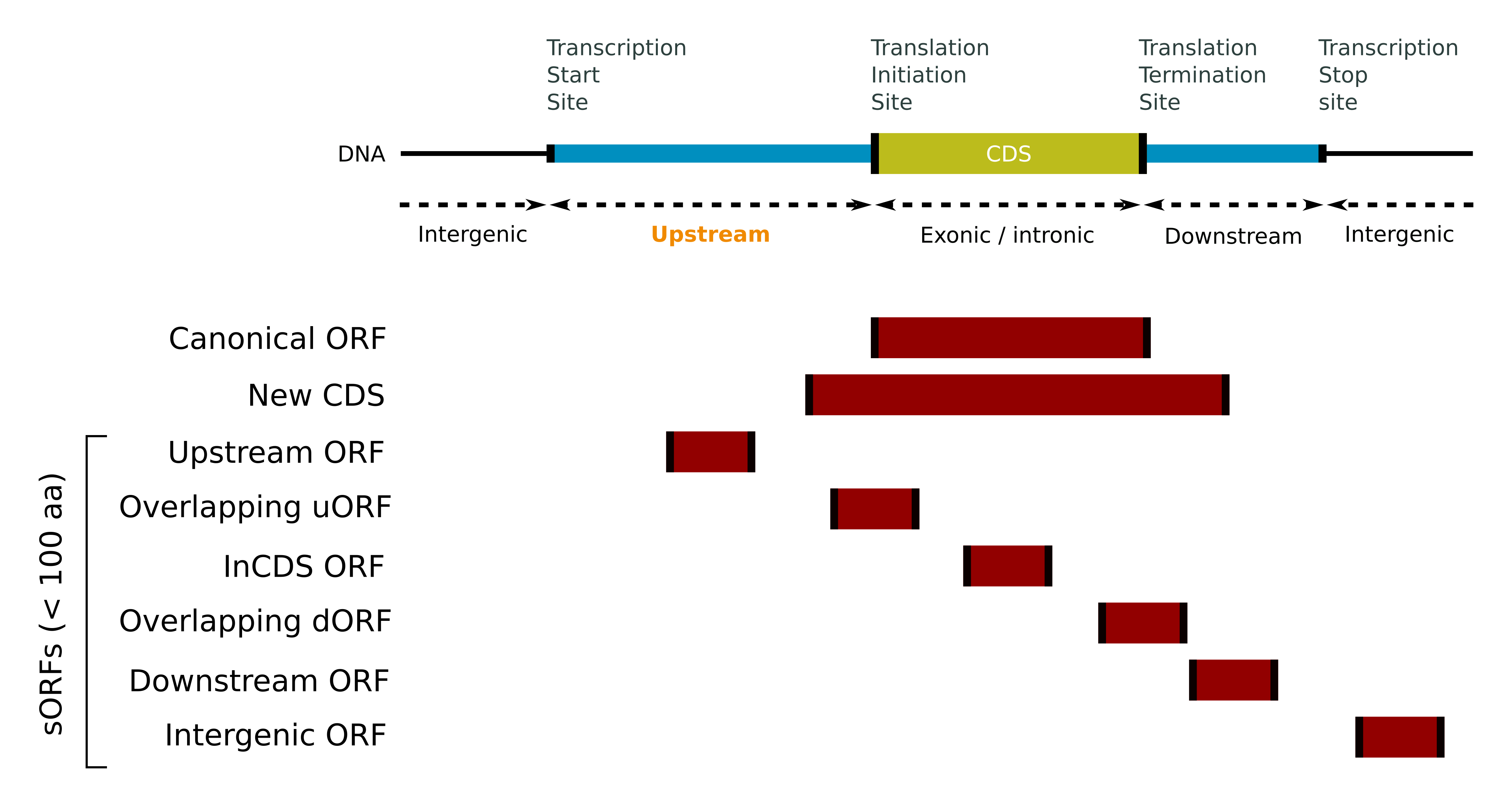
- ORF annotations based on length
- sORF (short ORF): The ORF sequence is shorter than 100 amino acids (stop codon and intron excluded).
- ORF annotations based on the transcript biotype
- Intergenic: The ORF is located on a 'Long intergenic ncRNA' or 'lincRNA' biotype.
- ncRNA: The ORF is located on a 'Non coding', 'ncRNA', 'Processed transcript', 'processed_transcript', 'Long non-coding RNA', 'lncRNA', '3' overlapping ncRNA', 'Macro lncRNA', 'Long intergenic ncRNA', 'lincRNA', 'miRNA', 'miscRNA', 'piRNA', 'rRNA', 'siRNA', 'snRNA', 'snoRNA', 'tRNA', or 'vaultRNA' biotype.
- Pseudogene: The ORF is located on a 'Pseudogene', 'IG pseudogene', 'Polymorphic pseudogene', 'Processed pseudogene', 'processed_pseudogene', 'Transcribed pseudogene', 'transcribed_processed_pseudogene', 'transcribed_unprocessed_pseudogene', 'unprocessed_pseudogene', 'transcribed_unitary_pseudogene', 'Translated pseudogene', 'Unitary pseudogene', 'unitary_pseudogene' or 'Unprocessed pseudogene' biotype.
- NMD: The ORF is located on a 'Non sense mediated decay', 'NMD', 'nonsense_mediated_decay' or 'non_stop_decay' biotype.
- Readthrough: The ORF is located on a 'Readthrough' or 'Stop codon readthrough' biotype.
- ORF annotations based on the relative position
- Upstream: The start codon of the ORF is located upstream of the CDS start codon and the stop codon of the ORF is located upstream of the CDS stop codon. Note that if both the start and the stop codons are the same for the ORF than the CDS, then the ORF is annotated CDS instead.
- Downstream:
- The start codon of the ORF is located downstream of the CDS start codon and the stop codon of the ORF is located downstream of the CDS stop codon. Note that if both the start and the stop codons are the same for the ORF than the CDS, then the ORF is annotated CDS instead, or
- The ORF is located on a '3'overlapping ncRNA' biotype.
- Overlapping:
- The start codon of the ORF is located upstream of the CDS start codon and the stop codon of the ORF is located downstream of the CDS start codon and upstream of the CDS stop codon, or
- The start codon of the ORF is located downstream of the CDS start codon and upstream of the CDS stop codon and the stop codon of the ORF is located downstream of the CDS stop codon or
- The ORF is located on an 'Antisense' biotype.
- Intronic: The ORF is located on a 'retained_intron', 'sense_intronic' or 'sense_overlapping' biotype.
- InCDS: The start codon of the ORF is located downstream of the CDS start codon and the stop codon of the ORF is located upstream of the CDS stop codon. Note that if both the start and the stop codons are the same for the ORF than the CDS, then the ORF is annotated CDS instead.
- CDS: The start codon of the ORF is the same than the CDS start codon and the stop codon of the ORF is the same than the CDS stop codon.
- NewCDS: The start codon of the ORF is located upstream of the CDS start codon and the stop codon of the ORF is located downstream of the CDS stop codon. Note that if both the start and the stop codons are the same for the ORF than the CDS, then the ORF is annotated CDS instead.
- ORF annotations based on the reading frame
- Alternative: The ORF start is located on a different frame than the CDS start codon (i.e. the distances in bp between the first nucleotide of the ORF start and the first nucleotide of the CDS start is not a multiple of three).
- ORF annotations based on the strand
- Opposite: The ORF is located on the opposite strand of its transcript.
Cell types
Original data sources do not use a common thesaurus or ontology to name the cell types (e.g. 'HFF' and 'Human Foreskin Fibroblast') or may use non-biological meaning names (e.g. sORFs.org provides the name of the original publication as a cell type). In order to provide an uniform informative naming, we manually recovered the name of the cell line, tissue or organ used in these datasets and defined an unique name to be used in MetamORF for each cell line, tissue or organ. In addition, when feasible, we recovered the ontology terms matching the cell line, tissue or organ in the following ontologies:
| Ontology | Full name | URL |
|---|---|---|
| EFO | Experimental Factor Ontology | https://www.ebi.ac.uk/ols/ontologies/efo |
| BAO | BioAssay Ontology | https://www.ebi.ac.uk/ols/ontologies/bao |
| BTO | The BRENDA Tissue Ontology (BTO) | https://www.ebi.ac.uk/ols/ontologies/bto |
| OMIT | Ontology for MIRNA Target | https://www.ebi.ac.uk/ols/ontologies/omit |
| CLO | CLO: Cell Line Ontology | https://www.ebi.ac.uk/ols/ontologies/clo |
| NCIT | NCI Thesaurus OBO Edition | https://www.ebi.ac.uk/ols/ontologies/ncit |
| HCAO | Human Cell Atlas Ontology | https://www.ebi.ac.uk/ols/ontologies/hcao |
| CL | Cell Ontology | https://www.ebi.ac.uk/ols/ontologies/cl |
| OBI | Ontology for Biomedical Investigations | https://www.ebi.ac.uk/ols/ontologies/obi |
| FMA | Foundational Model of Anatomy Ontology | https://www.ebi.ac.uk/ols/ontologies/fma |
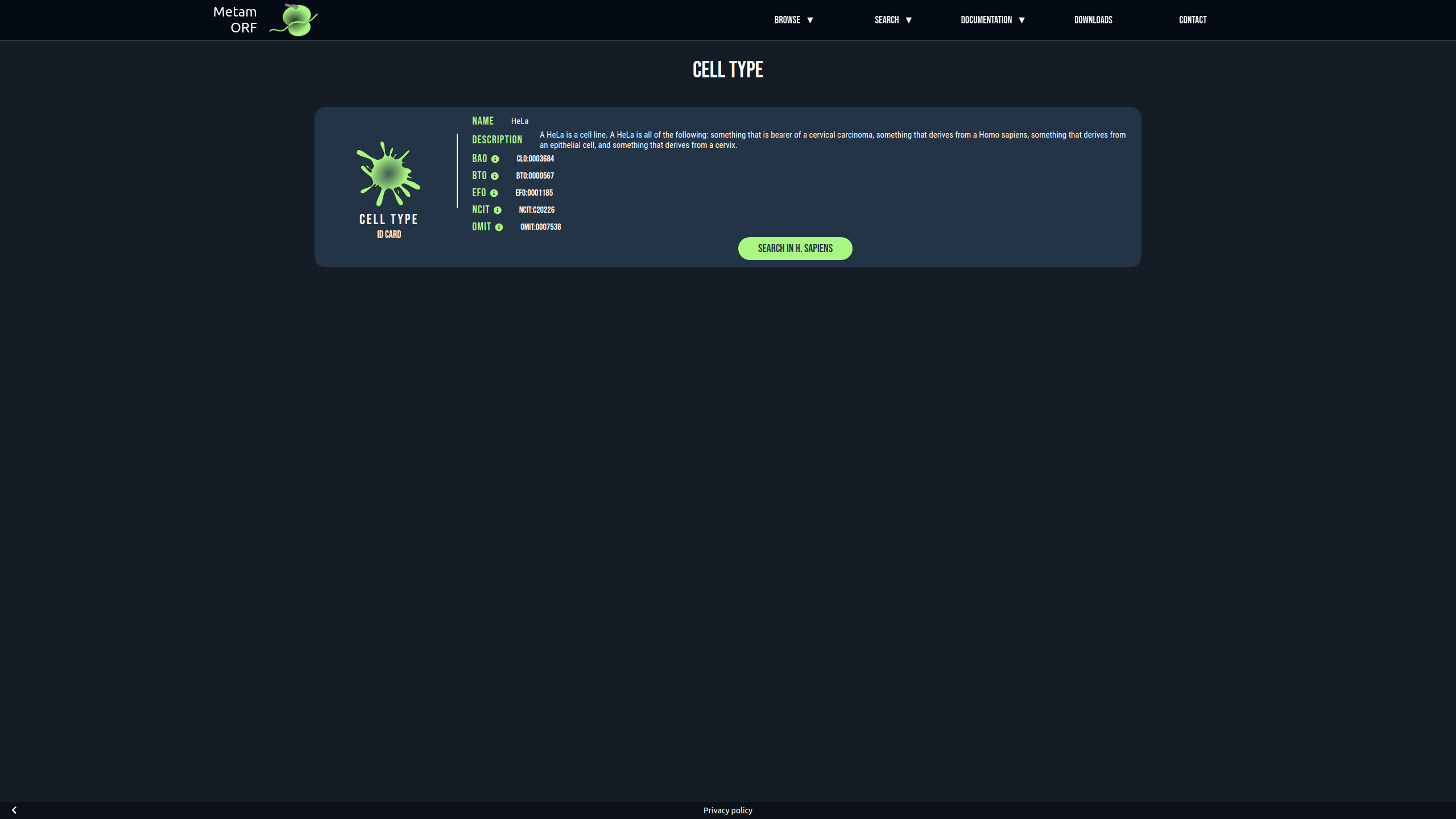
All the cell types existing in MetamORF may be accessed through the cell type browser. The pages of the cell types provide a short description of the cell type as well as the ontology terms matching with it. From these pages, it is also possible to browse all the ORFs identified in the cell type in H.sapiens and/or M.musculus.
Example of the page describing the HeLa cells
Kozak contexts definition
Kozak contexts have been assessed using the criteria defined by Hernandez et al., Trends in Biochemical Sciences, 2019 (doi: 10.1016/j.tibs.2019.07.001). To annotate the Kozak contexts, the nucleotides located from the -6 to +4 positions around the start codon (where +1 position correspond to the position of the first nucleotide of the start codon) have been downloaded from Ensembl for each existing ORF - transcript association. Then, the start flanking sequence has been compared to four patterns defining optimal, strong, moderate and weak Kozak contexts.
Kozak contexts have been assessed even for the ORFs using alternative start codons (i.e. start codons different from ATG), looking for the same patterns around the start codon. Hence, if you are willing to display exclusively the ORFs with 'strict' optimal Kozak context, you must use the 'optimal' filter and the 'ATG' start codon filter at the same time.
The following patterns have been used to identify the start codons:
| -6 | -5 | -4 | -3 | -2 | -1 | +1 | +2 | +3 | +4 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Optimal | G | C | C | R | C | C | N | N | N | G |
| Strong | N | N | N | R | N | N | N | N | N | G |
| Moderate | N | N | N | R | N | N | N | N | N | A/T/C |
| N | N | N | Y | N | N | N | N | N | G | |
| Weak | N | N | N | Y | N | N | N | N | N | A/T/C |
With R = A/G (purine), Y = C/T (pyrimidine).
From a technical point of view, the following regular expression have been used to perform the Kozak context assessment:
| Kozak context | Regex |
|---|---|
| Optimal | GCC[AG]CC.{3}G |
| Strong | .{3}[AG].{2}.{3}G |
| Moderate | (.{3}[AG].{2}.{3}[ATC]|.{3}[CT].{2}.{3}G) |
| Weak | .{3}[CT].{2}.{3}[ACT] |
Build URLs to MetamORF
If you are willing to include hyperlinks to MetamORF in your documents (reports, website...), you may build the URLs using the following rules:
- To point out to a gene page, use http://metamorf.hb.univ-amu.fr/gene/{species}/{id}, where {species} is the name of the species (either hsapiens or mmusculus), and {id} is the gene ID, symbol or alias.
- To point out to a transcript page, use http://metamorf.hb.univ-amu.fr/transcript/{species}/{id}, where {species} is the name of the species (either hsapiens or mmusculus), and {id} is the transcript official ID, name or MetamORF transcript ID.
- To point out to an ORF page, use http://metamorf.hb.univ-amu.fr/ORF/{species}/{id}, where {species} is the name of the species (either hsapiens or mmusculus), and {id} is the MetamORF ORF ID.
- To point out to a locus page, use http://metamorf.hb.univ-amu.fr/locus/{species}/{chr}:{start}-{end}/{strand}, where {species} is the name of the species (either hsapiens or mmusculus), {chr} is the name of the chromosome (with the 'chr' prefix), {start} and {end} are respectively the lower and upper bound of the locus of interest. The {strand} parameter is optional and may take as value '+', '-' or 'NA'.
Database schema
Database schema are available on request, please contact us if you need them.
Statistical description of the databases
Advanced statistical description of the databases are available on request at the .tar.gz format. The archive contains both html and png files generated with R and Rmd. Please be aware that these files includes technical information and a good knowledge of the database structure is required to interpret all the statistics described there. Please contact us if you are willing to get this archive.
RqueryORF package
A R package has been implemented to handle connections to the database and build some data frames summurazing the information contained in MetamORF. This package intends to be used to perform large queries on the databases and requires a good knowledge of the structure we used.
As this package is still under development, it is not yet available on the CRAN nor GitHub. Large queries cannot yet be performed through our web interface, so please feel free to contact us if you need to perform such queries.
Workflow used to build the database
Please see our publications for detailed material and methods. If you need information regarding technical details related to the build of the database and source code implemented, please contact us.
Source code
The source code used to build the database and its documentation are available on GitHub.
Get the full database
You may download the content of the full database at FASTA and BED formats on this page. If you are willing to get a copy of the full database at MySQL format, please contact us.
MetamORF softwares and languages
The pipeline used to build MetamORF has been developed using Python (v2.7) with SQLAlchemy ORM (sqlalchemy.org, v1.3.5). The database has been handled using MySQL (mysql.com, v8.0.16). Docker (docker.com, v18.09.3) and Singularity (singularity.lbl.gov, v2.5.1) environments have been used in order to ensure reproducibility and to facilitate deployment on high-performance clusters (HPCs).
The current web interface has been developed using the Laravel (laravel.com, v7.14.1) framework with PHP (v7.3.0), JavaScript 9, HTML 5 and CSS 3. NGINX (v1.17.10) web server and PHP server (v7.3.0) were deployed with Docker (docker.com, v18.09.3) and Docker-compose (v1.24.0) to ensure stability.
Web browser compatibility
The web interfaced has been tested with the following browsers.
| OS | Version | Screen resolution | Chrome | Firefox | Safari | Microsoft Edge |
|---|---|---|---|---|---|---|
| Linux | Ubuntu 16.04 | 1920 x 1080 | n/a | 86.0 | n/a | n/a |
| Linux | Ubuntu 18.04 | 2560 x 1440 | 78.0.3904.108 | 78.0.2 | n/a | n/a |
| Windows | 7 | 1440 x 1080 | 78.0 | 78.0 | n/a | n/a |
Database releases
- 1.0: First release of the database. Published on 01/07/2020.
Genome tracks and track hubs
Track hubs have been implemented in order to allow advance vizualisation of MetamORF data on the UCSC and Ensembl genome browsers.
UCSC Genome Browser
Public hub
MetamORF is not yet accessible through UCSC public hubs! Please use public sessions or MetamORF track hub URL instead. The documentation will be updated as soon as it becomes available.
The easiest way to use MetamORF genome track with the UCSC Genome Browser is to connect it from the Public Hub. To do this, go to the UCSC Hub Connect page and connect the appropriate MetamORF genome track you are willing to use.
Connect to MetamORF public hub with UCSC Genome Browser

Alternatively, you may also connect the Hub by yourself providing the url http://metamorf.hb.univ-amu.fr/hubDirectory/hub.txt.
Public sessions
UCSC public sessions allow users to save snapshots of the Genome Browsers and its current configuration, including the displayed tracks, positions etc. The sessions tools are convenient to easily share sessions with your collaborators. We take advantage of the Public Session tool to share with the rest of the community some MetamORF sessions we find interesting. Public sessions can be accessed directly at https://genome.ucsc.edu/cgi-bin/hgPublicSessions and search for MetamORF public session.
UCSC public sessions
Add custom track to the UCSC Genome Browser
Ensembl Track Hub
Public hub
MetamORF is not yet accessible through Ensembl public hubs! Please use MetamORF track hub URL instead. The documentation will be updated as soon as it becomes available.
The easiest way to use MetamORF genome track with the Ensembl Genome Browser (e!) is to connect it from the Public Hub. To do this, go to https://www.ensembl.org/info/website/tutorials/userdata.html and select Custom track. In the window that is open, select Track Hub registry search and search for MetamORF tracks. Then, you just need to attach the appropriate Hub.
Connect to MetamORF public hub in e!
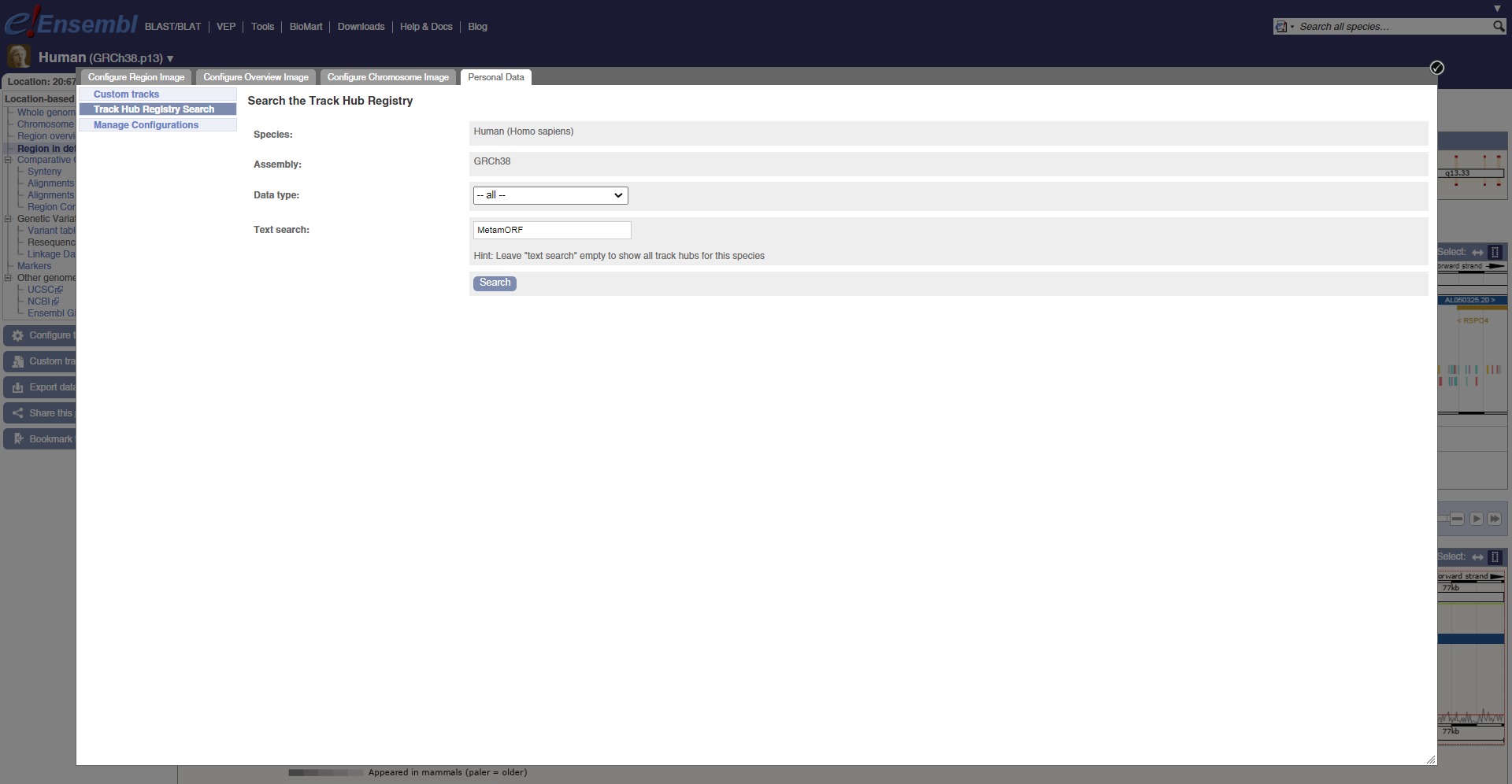
Alternatively, you may also connect the Hub by yourself providing the url http://metamorf.hb.univ-amu.fr/hubDirectory/hub.txt.
Add custom track in e!
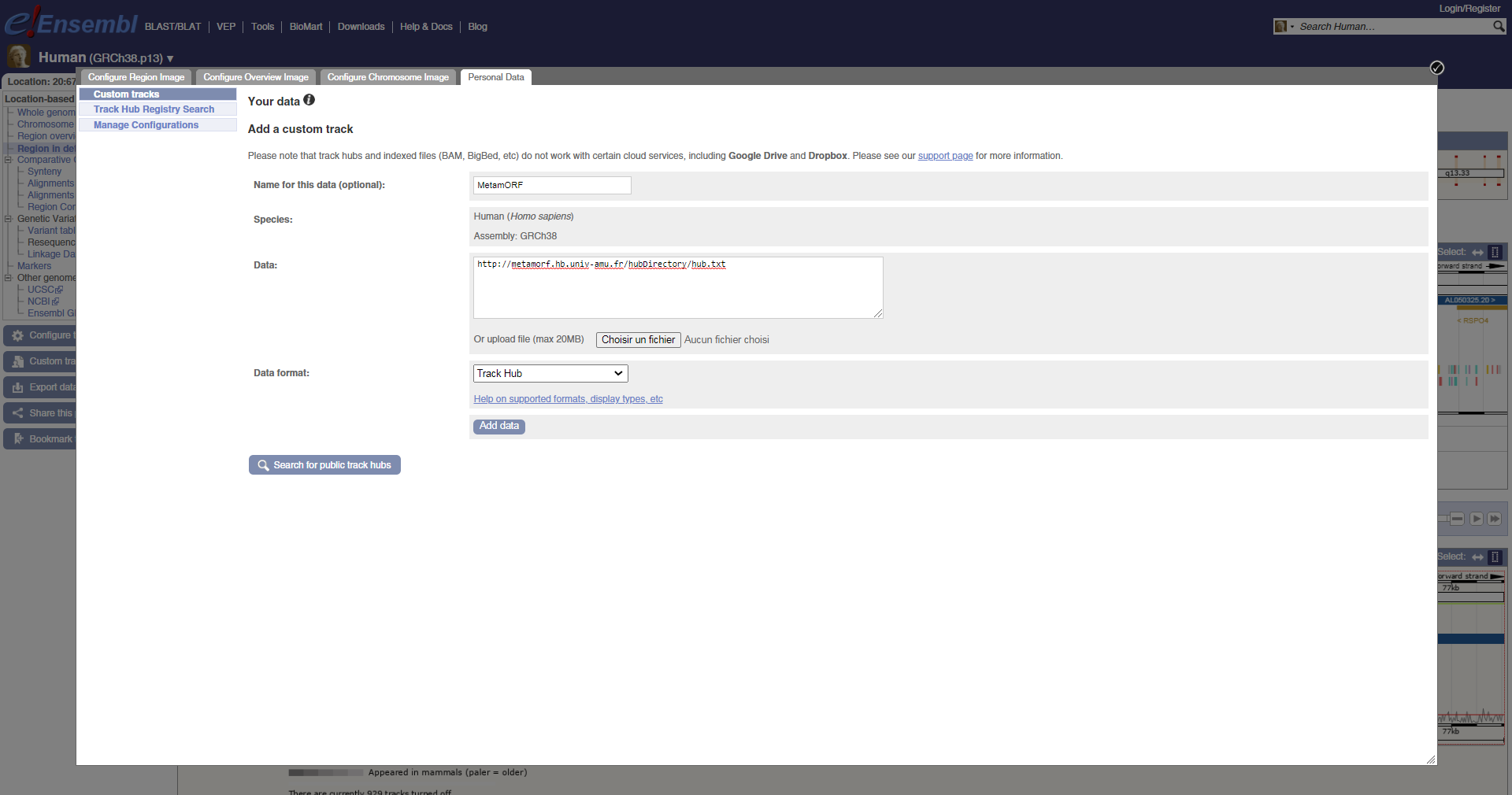
New to Track Hubs?
If you do not kown what a Track Hub is, please check the UCSC Track Hub documentation. Briefly, Track Hubs are web-accessible directories of genomic data that can be viewed on Genome Browsers. It offers a convenient way to view and share very large sets of data. The track hub utility allows efficient access to data sets from around the world through the familiar Genome Browser interfaces.
We advice to read the UCSC Track Hub paper, Raney BJ, et al. Track Data Hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser. Bioinformatics. 2014 Apr 1;30(7):1003-5.
If Ensembl is your favorite Genome Browser, you may find a tutorial here.
MetamORF Genome tracks and Track Hub
MetamORF Track Hub URL: http://metamorf.hb.univ-amu.fr/hubDirectory/hub.txt.
Track Hubs may sometimes experience limitations on large regions due to the high volume of data. In such cases, it may be interesting to download yourself MetamORF BED files and upload them on your favorite Genome Browser (UCSC, IGV, e!...). To download these files, please check the downloads page.
Frequently Asked Questions
- In my results, there is a transcript which official ID is UNKNOWN TRANSCRIPT, what does that mean?
- Why does my gene has two aliases nearly identical, with one of them having a OFF: prefix?
- Why I am not able to find any gene when I search using a HGNC or a NCBI ID?
- My ORF seems to be related to several genes. How is that possible?
- The transcript I am looking seems to be related to another gene than the one described on Ensembl or NCBI website. Which database should I trust?
- It seems that the ORF I am looking at is related to a gene with an unusual name. What happened?
- How are registered mitochondrial and sexual chromosomes in MetamORF?
- Which genome annotations are you using in your database?
- I am confused about the start and stop (or end) coordinates? Why are stop and end coordinates always higher than the start coordinates, even for ORFs or transcripts located on the reverse strand?
- To which nucleotides are actually corresponding the start, stop and end positions?
- I am confused about the absolute coordinates? What coordinate counting system do you use?
- What are the difference between absolute and relative coordinates? Why are the relative coordinates sometimes missing? Which ones should I use?
- What is the difference between an ORF and a CDS?
- What are the nucleotides included in the flanking sequences around the start codon?
- According to you MetamORF contain unique entries. Still, when I am looking at data in a genome browser it seems that a lot of ORFs are similar. How is that possible?
- I have data about sORFs that might be of interest for your database. How can I submit data to be included in MetamORF?
- MetamORF web interface is not working fine. I found information that seems to be inaccurate in your database. I have a question that is not treated in you FAQ.
- How should I cite MetamORF?
In my results, there is a transcript which official ID is UNKNOWN TRANSCRIPT, what does that mean?
When we integrated the data from the data sources, information about the transcript were sometimes missing, either for the whole source or only for some of the ORFs. In such cases the ORF was considered as located on an 'unknown' transcript. Hence all ORFs related to the same gene and for which transcript is unknown are grouped together on the a transcript with the UNKNOWN TRANSCRIPT ID.
Each of the 'unknown transcripts' have a different MetamORF unique ID as soon as they are related to different genes. Note that searching for 'unknown transcripts' from the search page is not allowed as there are two many transcript with this ID in our database. We suggest to use one of the other search mode (gene, locus, ORF) and then click on this transcript if you need to access to the transcript page or to use its MetamORF ID to perform the search.
By definition, these transcripts are obviously missing all information (start, end, strand, CDS...) and any ORF located on a known transcript will not appear on the unknown transcript.
Why does my gene has two aliases nearly identical, with one of them having a OFF: prefix?
During the build of MetamORF, we started by importing the cross-references from HGNC, MGI, NCBI and/or Ensembl databases. Genes usually have an alias considered as official (e.g. the official alias of 'GADD34' is 'PPP1R15A'). The alias known as the official alias is preceeded by the OFF: prefix in order to be identified more easily.
Why I am not able to find any gene when I search using a HGNC or a NCBI ID?
When performing you search, HGNC IDs must be preceed by the HGNC: prefix and NCBI IDs must be preceed by the NCBI: prefix, in order to avoid any confusion in external references. As Ensembl ID can be easily recognized, this is not necessary to add any prefix to them. Symbols, names and aliases being idependent to these resources, they have to be provided without any prefix.
My ORF seems to be related to several genes. How is that possible?
In ORF pages, it may happen that an ORF is related to several genes. This may happen when the same ORF was reported by two different data sources which were using different gene ID, name or alias, that are supposed to design the same gene but unknown by MetamORF. Because one of the cross-reference was missing from MetamORF, it was not able to detect that the gene was actually the same, and the ORF has thus been related to distinct genes.
The transcript I am looking seems to be related to another gene than the one described on Ensembl or NCBI website. Which database should I trust?
When integrating data from the original sources, MetamORF kept the relationship between the transcripts and the gene provided by the data source. Hence, if for some reason the relationship provided by the data source was not accurate, the relationship registered in MetamORF may be wrong, and a transcript may have been inaccurately associated to a gene.
In addition, when the transcript information was provided but the gene ID was missing, MetamORF tried to recover the gene information from the transcript ID by querying Ensembl database. If for some reason, the gene name recovered was inaccurate (due to versionning issues for instance), then again the relationship registered in MetamORF can be wrong.
Finally, it may happen that some gene aliases are refering to two distinct genes (e.g. RUFY3 refers to both 'RUFY3' and 'FYCO1' genes). When this happened, MetamORF tried to look for the gene which had the provided alias as official alias (in this example, it would have associated RUFY3 to the actual RUFY3 gene, i.e. the gene with Ensembl ID 'ENSG00000018189'). If the transcript was actually related to the other gene (in this example, 'FYCO1'), then MetamORF will associate it by mistake with the wrong gene ('RUFY3' in our example).
Hence, due to these various issues, it may be highly difficult to find the origin of the bad association. We suggest in such cases to trust the Ensembl or NCBI databases as the mistake is susceptible to have been introduced during the processing of data, either by the original data source or by MetamORF workflow.
It seems that the ORF I am looking at is related to a gene with an unusual name. What happened?
Sometimes, information about the gene was missing from the data source. In such cases, we tried to recover it from the transcript ID, or (when it was not provided) from the ORF coordinates. It may happen that the ORF coordinates were found ovelapping with several genes on the same strand or no gene. In such case the ORF was registered as related with an 'unknown' genes. Contrary to 'unknown' transcripts, 'unknown' genes are using different names. The following nomenclature is used:
- OVERLAPPING_GENES_gene1_gene2 gene ID is used for ORFs located in a locus overlapping exactly two genes (gene1 and gene2).
- OVERLAPPING_GENES_gene1_to_gene2 gene ID is used for ORFs located in a locus overlapping at least three genes (the name of the two genes are the one located at extremal positions; genes located between them are omitted).
- OVERLAPPING_LNCRNAS_gene1_gene2 gene ID is used for ORFs located in a locus overlapping with no protein coding gene but overlapping exactly two non coding genes (gene1 and gene2).
- OVERLAPPING_LNCRNAs_gene1_to_gene2 gene ID is used for ORFs located in a locus overlapping with no protein coding gene but overlapping with at least three non coding genes (the name of the two genes are the one located at extremal positions; genes located between them are omitted).
- INTERGENIC_GENE_chrName gene ID is used for ORFs located in a locus of the chromosome chrName, where there is not any known gene.
- CONFLICT_GENE_gene1_gene2_chrName gene ID is used for ORFs located on the chromosome chrName and on a known transcript but for which conflicting information regarding the gene were provided in the origial data source. This happen when the same data source is associating a particular transcript ID with two distinct gene IDs. This may happen when (i) the two gene IDs are actually refering to the same gene but the cross-reference was unknown by MetamORF, (ii) the two gene IDs are actually distinct genes (in such case the mistake comes from the original data source and MetamORF was not able to solve the issue).
How are registered mitochondrial and sexual chromosomes in MetamORF?
The ORFs (or genes) located on mitochondrial chromosomes are registered as located on the MT chromosome.
The ORFs (or genes) located on a specific sexual chromosome (i.e. either X or Y), are defined using the appropriate chromosome name (X or Y). The ORFs (or genes) indifferently located on the X or Y chromosome are registered as located on the X|Y chromosome.
The chromosome names are saved without the 'chr' prefix in the MetamORF database.
Which genome annotations are you using in your database?
All coordinates are using GRCh38 (hg38, Ensembl release 90) genome annotations for H. sapiens and GRCm38 (mm10, Ensembl release 90) genome annotations for M. musculus.
I am confused about the start and stop (or end) coordinates? Why are stop and end coordinates always higher than the start coordinates, even for ORFs or transcripts located on the reverse strand?
When it comes to provide start and stop (or end) coordinates, to improve programming efficiency, it is easier to consider the start coordinates as the lower bound of the genomic area considered, and the stop (or end) as the higher bound of the genomic area; whatever it actually corresponds to the start or the stop (or end) coordinates.
Hence, this is the strand that will give you information about the actual start and stop (or end) of the ORF, CDS or transcript. For instance, the position of the start codon will be registered in the start attribute for an ORF located on the forward strand whilst the position of its stop codon will be registered in the stop attribute.
On the contrary, the position of the start codon will be registered in the stop attribute for an ORF located on the reverse strand whilst the position of its start codon will be registered in the start attribute.
Be carefull, the coordinates of the ORF exons do not follow this convention. On the contrary, they are provided using their order on the transcript, meaning that the first coordinate will always correspond to the coordinates of the first nucleotide of the first codon of the exon. Be also aware that on ORF pages, the coordinates are registered by exon (the first coordinates corresponds to its start whilst the second corresponds to its end). In the exported CSV files, exon start and ends are registered in two different columns. Please see the documentation related to the ORF page and related to the CSV exports for more information.
To which nucleotides are actually corresponding the start, stop and end positions?
The start position is always providing the coordinates of the first nucleotide of the feature, i.e. either the location of the first nucleotide of the start codon of an ORF, or the location of the first nucleotide of the exon of an ORF, or the location of the first nucleotide of the start codon of an CDS, or the location of the first nucleotide of the transcript.
The stop positions is always providing the coordinates of the last nucleotide of the feature, i.e. either the location of the last nucleotide of the stop codon of an ORF, or the location of the last nucleotide of the exon of an ORF, or the location of the last nucleotide of the stop codon of an CDS, or the location of the last nucleotide of the transcript.
Keep in mind that start and stop (end) position may be reversed for features located on the reverse strand. See the previous question for more information regarding this issue.
I am confused about the absolute coordinates? What coordinate counting system do you use?
MetamORF web interface is displaying both the start and end coordinates of features in a 1-based sytem (fully-close system, also sometimes inaccuratelly called 1-based start, 1-based end system). The coordinates are stored in our database using the same system and coordinates exported in CSV and FASTA files are also using a 1-based coordinate system.
Caution: In order to respect the usual conventions, the coordinates exported in BED file are saved in a 0-based start, half-open (also inaccuratelly called 0-based start, 1-based end) system.
If you are confused about these definitions, we strongly advice to read this article published on the UCSC blog which provides detailed information regarding the coordinate counting systems.
What are the difference between absolute and relative coordinates? Why are the relative coordinates sometimes missing? Which ones should I use?
Absolute coordinates are providing the coordinates on the chromosome, in a 1-based (fully-close) system, meaning that the first nucleotide of the chromosome is numbered 1. Using absolute coordinates allows to locate features anywhere on the chromosome, including in intergenic regions or intron. Nevertheless, because of intron excision during the splicing (and alternative splicing) two features located in distant places on the chromosome can be actually near to each other on the transcript. Hence, relative coordinates provide the location on the features on the transcript. Because we are sometimes missing information about the transcript, it is thus impossible to compute relative coordinates. Moreover as relative coordinates are defined relatively to the transcript, you will always find them related to a particular transcript (and relative coordinates may vary for ORFs that are expressed on different transcripts).
Absolute coordinates are provided by the data sources and lift overed to the last annotation version (i.e. converted from their original annotation to the current one), whilst relative coordinates are computed using the R ensembldb and annotationHub packages. When possible and if you are working on the genome, we advice to use absolute coordinates as they went over less computational steps and misconversion may sometimes happen. Nevertheless, if you are working on transcripts, the relative coordinates are more relevant and should more likely be prefered.
What is the difference between an ORF and a CDS?
Open Reading Frames (ORFs) are usually defined as sequences delimited by a start codon and a stop codon and potentially translatable into functional peptides or proteins.
Coding DNA sequences (CDSs) are usually defined as ORFs actually translated into functional proteins. For more clarity, we considere here the CDSs as the canonical sequences of the transcript actually encoding a functional protein.
Hence, ORFs registered in MetamORF are newly identified ORFs and many ORFs may be located on the same transcript, whilst there is at most one CDS on a transcript. Our algorithm relies on these definition to annotate the ORFs. See the ORF annotation section of this documentation for more information regarding the nomenclature we used to define the ORFs.
What are the nucleotides included in the flanking sequences around the start codon?
When the transcript of an ORF was known and the relative coordinates of the ORF susccessfully computed, we tried to recover the sequences around the start codon of the ORF (by querying Ensembl databases). The nucleotides from the -6 position to the +1 position were included in this sequence, where the +1 position correspond to the first nucleotide of the start codon. Hence, this sequence is always containing exactly 10 nucleotides. The flanking sequences were used to compute the Kozak context.
e.g. If the flanking sequence is 'GCCACCATGG', then the ORF has ATG as start codon (+1 to +3 positions), GCCACC as sequence upstream of the start codon (-6 to -1 positions) and G as sequence downstream of the start codon (+4 position).
According to you MetamORF contain unique entries. Still, when I am looking at data in a genome browser it seems that a lot of ORFs are similar. How is that possible?
Two ORFs are considered as identical if they have:
- The same chromosome name
- The same strand
- The same genomics coordinate for the start position
- The same genomics coordinate for the stop position
- The same number of exons
- The same genomics coordinates for the start and end of each exon
Hence, two ORFs that have at least one of this attribute which varies will be considered as different and registered as two distinct entries (with two different IDs) in MetamORF. It may happen that two ORFs share a lot of common information and only differ on one of these attributes (e.g. same features except for the start codons which are located at two position spaced of 6 nucleotides), in which case they will look very similar on genome browsers, still they are actually different (according to this definition).
I have data about sORFs that might be of interest for your database. How can I submit data to be included in MetamORF?
If you have data describing new sORFs, we would be happy to integrate them in our database. Nevertheless, the processing of data by MetamORF is computationally heavy and integration of new data cannot be fully automatized. So please contact us to discuss about this.
In order to facilitate the integration of new data in MetamORF, please note that:
- Data should specifically be describing short open reading frames (sORFs),
- Both computational predictions and experimental data will be considered for inclusion,
- Data describing canonical open reading frames will not be considered for inclusion,
- Data sources already included (see previous section of the documentation), in particular being part of sORFs.org database (and already inserted in MetamORF), will not be considered for inclusion,
- You may find here guidelines regarding the format we expect to facilitate the integration of your data in MetamORF. Fell free to contact us if you have any question.
MetamORF web interface is not working fine. I found information that seems to be inaccurate in your database. I have a question that is not treated in you FAQ.
If you are facing one of this issue, please contact us so we can help you and ensure there is no bugs in MetamORF or fix them. We tried to address about the most frequently asked question to complete the documentation, some of them may be missing, so please contact us when you have a question so we can fill it!
How should I cite MetamORF?